
Abstract
Unprecedented advances have been made in the treatment of cancer through the use of immune checkpoint blockade, with approval of several checkpoint blockade regimens spanning multiple cancer types. However, responses to this form of therapy are not universal, and insights are clearly needed to identify optimal biomarkers of response and to combat mechanisms of therapeutic resistance. A working knowledge of the hallmarks of cancer yields insight into responses to immune checkpoint blockade, although the focus of this is rather tumour-centric and additional factors are pertinent, including host immunity and environmental influences. Herein, we describe the foundation for pillars and hallmarks of response to immune checkpoint blockade, with a discussion of their relevance to immune monitoring and mechanisms of resistance. Evolution of this understanding will ultimately help guide treatment strategies to enhance therapeutic responses.
Similar content being viewed by others
Introduction
The treatment of cancer has been arduous, marked by just as much heterogeneity in cancer treatment modality and outcome as is now known to exist at a cellular and molecular level within tumours themselves. Cancer burden, morbidity, and mortality have a wide reaching impact globally, but recent advancements in precision medicine have given the field of oncology an opportunity to greatly improve therapeutic strategies. As the relative ‘new kid on the block’, cancer immunotherapy differs from conventional chemotherapeutic agents in that its mechanism of action employs, engages, or enhances a functional immune response to tumour cells, rather than aiming principally to physically remove or destroy cancer cells through inherent radio- or chemical toxicity. Importantly, although immunotherapy is commonly thought of as a new treatment modality, the first immunotherapy approaches in fact predate the discovery and development of cytotoxic agents for the treatment of cancer, or even the discovery of X-rays, let alone the therapeutic use of non-ionising radiation. Furthermore, immunotherapy encompasses several subtypes of treatment modality, including vaccination strategies, cell-based therapies using the patient’s own immune cells with or without ex vivo modification, and immunomodulatory agents, of which checkpoint inhibitor therapies have been the most broadly successful to date.
Physiologic role and therapeutic targeting of immune checkpoints
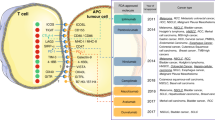
Unopposed immune activation can be at least as damaging as an ineffective response, necessitating a dynamic system of regulatory signals to integrate the prevailing immune stimuli and direct immune responses appropriately. Initial immune activation requires recognition of the target, which itself is a multistep process classically requiring antigen expression by tumour cells, and its processing and presentation to helper T cells by specialised antigen presenting cells (APCs, e.g., dendritic cells) in the context of class II human leukocyte antigen (HLA; Figure 1). Whether a cognate HLA/antigen – T-cell receptor interaction results in T-cell proliferation and activation is determined by the presence of additional co-stimulatory signals, principally delivered by the engagement of CD28 on the T cell by CD80/86 on the APC (Figure 1). Without this vital second signal, the interaction may be biologically interpreted as representing recognition of a non-pathogenic (or ‘self’) antigenic stimulus to which tolerance may develop. However, in the presence of appropriate co-stimulation, an active immune response against the inciting antigen can proceed, with the generation of humoral responses, recruitment of a cytotoxic T-cell response (HLA class I-restricted) and release of numerous cytokines necessary for effector cell proliferation, survival, localisation, and effector function. Many other stimulatory signals are active throughout the immune response phase, including inducible T-cell co-stimulator (ICOS), glucocorticoid-induced TNFR-related protein, and tumour necrosis factor receptor superfamily members 4 (OX40 or CD134) and 9 (4-1BB or CD137), which function in the amplification and maintenance of overall immune activation (Figure 1).

The cellular immune response to cancer is complex and involves a diverse repertoire of immunoregulatory interactions principally involving antigen presenting cells (APC), T cells, and tumour cells. Presentation of distinct antigen epitopes to CD8+ and CD4+ T cells in the context of major histocompatibility complex class I (on APC or tumour cells directly) and class II (on APCs), respectively, facilitates tumour cell recognition, but numerous other molecular interactions (inset boxes) and input from paracrine and humoral factors (cytokines/chemokines, shown with arrowed lines) integrate to determine the ultimate outcome of immune recognition. Elaboration of survival and inflammatory cytokines, such as IL-2 and IFN-γ, can promote a cytotoxic (CD8+) T-cell response with consequent tumour-directed lytic activity mediated by release of cytotoxic granule contents (e.g., perforin and granyzme) as well as triggering of apoptotic pathways by tumouricidal cytokines (e.g., TNF-α and IFN-γ) and death receptor ligation (e.g., FAS:FAS-L). Debris released from apoptotic/necrotic tumour cells may be taken up by APC and presented in a cycle of immunogenic cell death. However, prolonged immune activation is adaptively opposed by upregulation of immunoinhibitory molecules (e.g., CTLA-4, PD-1, TIM3, TIGIT, and CTLA-4), or their ligands, many of which may be subverted by tumour cells in order to escape immune attack. Release of anti-inflammatory, immunoregulatory or Th2-skewed cytokines also contributes to dampening of the cellular response.
To achieve immune homeostasis, numerous negative feedback stimuli act to dampen the immune response, including the well-described negative regulatory molecules cytotoxic T-lymphocyte-associated protein 4 (CTLA-4 or CD152) and programmed death 1 (PD-1 or CD279). Cytotoxic T-lymphocyte-associated protein 4 is expressed on the T-cell surface and competes with CD28 for binding to CD80/86, providing an inhibitory stimulus upon engagement (Figure 1). It is thought that the action of CTLA-4 may be most relevant at the T-cell priming stage in regional secondary lymphoid organs, thus ultimately acting to impair T-cell help and the generation of an effector T-cell population and its egress back into the tumour. Programmed death 1 is a T-cell surface receptor that delivers inhibitory signals upon engagement with its ligands PD-L1/2, and these ligands may be upregulated in the setting of high levels of IFN-γ (termed adaptive immune resistance), but may also be expressed in the tumour microenvironment via oncogenic expression on tumour cells or expression on other stromal elements (Figure 1) (Pardoll, 2012). Programmed death 1 expressing T cells are thought to represent populations that have largely ‘seen’ their antigen in situ (i.e., within the tumour) and are thus considered a more tumour-specific population than T cells arrested at the priming stage by CTLA-4, however, high levels of PD-1 are also associated with an ‘exhausted’ T-cell phenotype (Wherry and Kurachi, 2015). Multiple other inhibitory ‘checkpoints’ have been identified, including lymphocyte activation gene 3 (LAG3 or CD223), and T-cell immunoglobulin 3 (TIM3) and T-cell immunoglobulin and ITIM domain (TIGIT), for which ligands expressed on tumour or stromal cells may act synchronously or sequentially to promote overall physiologic suppression of immune responses (Figure 1).
Elucidation of the complex web of stimulatory and inhibitory signals that contribute to the tug-of-war of immune regulation and their dysregulation in cancer presents clear therapeutic opportunities targeting these to enhance anti-tumour immune responses. The impressive proof-of-principle for this approach came with the report in 2010 of a phase III clinical study of CTLA-4 blockade with the monoclonal antibody ipilimumab in patients with metastatic melanoma, which demonstrated enhanced survival in treated patients (Hodi et al, 2010). Although objective responses were infrequent (<11%), checkpoint inhibitor therapies as a class have been characterised by durability of responses in those patients who achieve an objective response, contributing to a notable ‘tail’ in the survival curve of long-term survivors. Importantly, while patient-level data are frequently limited, even summary data indicate that objective responses are not an absolute requirement for a survival benefit from ipilimumab. In the last six years, four engineered monoclonal antibody immune checkpoint inhibitor agents have been approved in more than 50 global markets for six forms of cancer; ipilimumab (anti-CTLA-4), pembrolizumab and nivolumab (anti-PD-1), and atezolizumab (anti-PD-L1), with response rates of up to 40–50% with PD-1-based therapy. Combination strategies, including immune checkpoint inhibitors, with different mechanisms of action have also been approved (anti-CTLA-4 and anti-PD-1) and are associated with higher response rates (exceeding 60%) though toxicity to therapy is a significant issue (Larkin et al, 2015; Postow et al, 2015). Although the greatest strength of combination regimens may lie in converting a proportion of patients destined not to benefit from single-agent checkpoint blockade into long-term survivors, reliable methods to identify these patients before therapy remain elusive. In addition, matured outcome data will be necessary to determine whether combination checkpoint blockade confers superior overall survival outcomes relative to monotherapy approaches such as PD-1 blockade alone.
Despite significant clinical gains in the setting of treatment with immune checkpoint blockade, limitations to this therapeutic strategy have inevitably surfaced as they have for prior generations of novel therapeutic strategies. Treatment with current checkpoint inhibitor monotherapy is not effective in all cancer types, as tumours with lower mutational burden and/or lower immunogenicity may be inherently resistant to this form of therapy. Even in the setting of initial responses in favourable tumour types, resistance may develop. This may be related to redundancy in the very web of activating and inhibitory molecules targeted by immune checkpoint inhibitors (Koyama et al, 2016), though other mechanisms of therapeutic resistance have also been identified including adaptive loss of antigenicity, recognition machinery, and transience of the inflamed tumour microenvironment. On top of this, strong predictive biomarkers of response to immune checkpoint blockade are currently lacking, and toxicity can be a major issue, particularly in combination strategies. All of these factors, as well as an appreciation of the cost of these agents and issues with access to therapy, call for a more comprehensive understanding of the hallmarks of response to immune checkpoint blockade in order to derive more tailored strategies.
Hallmarks of response to immune checkpoint blockade
There is a growing appreciation of the key factors contributing to response and resistance to immune checkpoint blockade, drawing upon features of the tumour itself (including the cancer genome, epigenome, and microenvironment), components of host immunity (both systemic and anti-tumour immunity), and external influences such as the microbiome (Figure 2). The ‘hallmarks of cancer’ described by Hanahan and Weinberg (2011) are tightly related to these responses, though current applications of the hallmarks are rather tumour cell centric. In contrast, a description of the hallmarks of response to immune checkpoint blockade must take into account more global features, recognising that tumours constitute a dynamic milieu and integrate numerous reinforcing and antagonistic signals from both local and systemic conditions. Herein, we describe the four ‘pillars’ and associated hallmarks of response to immune checkpoint blockade, with intimate interactions also noted between each of the pillars (Figure 2).

The core pillars and thematic hallmarks of anti-tumour immunity governing response to immune checkpoint blockade. Extensive research has identified numerous tumour-centric domains (shown in blue), including both static (existing genomic aberrations) and dynamic (epigenomic, metabolic and microenvironmental) features, which moderate anti-tumour immune responses and have impact on the efficacy of immune checkpoint blockade. Relevant metrics of overall immunocompetence, and systemic factors regulating the balance between immunotolerant and inflammatory states (e.g., innate and adaptive cell abundance and composition, immune cell circulation/sequestration, cytokine levels; shown in brown) are gradually being quantified. Environmental factors previously not implicated in directly modulating the anti-tumour response are now recognised to impact on immune checkpoint response (shown in green); principal among these sources of immunomodulation is the gut microbiota, while environmental stresses (e.g., thermal stress) and other tumour-remote immune-pathogen interactions may produce humoral factors that impact upon the specific anti-tumour response.
Tumour genome and epigenome
We have gained a tremendous amount of information on cancer genomics over the past few decades through the use of next-generation sequencing techniques, which has helped to usher in the age of precision medicine, although how best to use this data in the clinic remains unclear.
Genomic alterations in cancer may have divergent roles—potentially enhancing anti-tumour immunity in some instances and conferring resistance in others. A prime example of how mutations may enhance responses comes from evidence that tumour types with higher average mutational loads (such as melanoma and non-small cell lung cancer) have a much higher response to treatment with immune checkpoint blockade than those with a lower mutational burden, likely related to a proportionally higher burden of immunogenic cancer-specific ‘neoantigens’ (Van Allen et al, 2015; McGranahan et al, 2016). In addition to this, subtypes of cancer with specific genomic alterations leading to increased mutational burden may also demonstrate enhanced responses to immune checkpoint blockade, such as microsatellite unstable colorectal cancers resulting from mutational loss or epigenetic silencing of DNA mismatch repair genes and resultant genomic instability (Le et al, 2015). Similarly, it has been noted that several mutagen exposures – such as UV light in melanoma, and tobacco smoke in non-small cell lung cancer – display strong co-associations with mutational burden and checkpoint blockade immunotherapy response (Rizvi et al, 2015). However, simply harbouring high mutational levels is not the complete story, as neoantigen proteins must be expressed, processed, and of sufficient binding characteristics in the context of HLA to be immunogenic although evidence suggests that the predictive value of neoantigen load is not driven by the small proportion of neoantigens with high predicted HLA-binding affinity (Van Allen et al, 2015). Detailed exomic analysis of a cohort of melanoma patients treated with CTLA-4 blockade revealed a shared repertoire of tetrapeptide neoantigen sequences in patients who derived clinical benefit; the immunogenicity of several neoantigen peptides was confirmed using patient-derived lymphocytes in vitro (Snyder et al, 2014). Importantly, the association with response was stronger for the neoantigen signature than for overall mutational burden, consistent with the notion that overall mutational burden increases the likelihood that a tumour is immunogenic, but that it may not be an absolute requirement for checkpoint blockade response. Importantly, a number of other types of antigens exist in cancer (cancer germline antigens, differentiation antigens, over-expressed antigens, and viral antigens), which can help to elicit anti-tumour immune responses (Blankenstein et al, 2012).
In contrast to the potentially pro-immunogenic impact of genomic alterations, there is a growing body of evidence regarding other genomic and epigenomic alterations in tumours that may impair immune responses and facilitate resistance to immune checkpoint blockade. Constitutive mitogen-activated protein kinase (MAPK) activation by mutations in the BRAF oncogene (and other MAPK pathway mutations) contributes to immune evasion by altering expression of tumour-associated antigens and major histocompatibility complex expression (Boni et al, 2010). Loss of expression of the tumour suppressor gene PTEN (either by mutations or copy number alterations) is also associated with impaired response to immune checkpoint blockade (Peng et al, 2016). Several studies have shown that immunotherapy resistance may originate in more than one compartment of the tumour microenvironment, with signals derived from tumour cells preventing immune infiltration (e.g., Wnt-β-catenin, PPAR-γ, FGFR3) while the dynamic interplay of anti-tumour immune attack adaptively moulds the landscape of immunomodulatory elements present over time (Spranger et al, 2013, 2015; Sweis et al, 2016). In line with evidence that interferon signatures play a significant role in the response to immune checkpoint blockade and may potentially act as clinical biomarkers (Ribas et al, 2015), JAK1/2 mutations have been identified in patients resistant to PD-1 blockade, acting via disruption of tumour-inhibitory interferon signalling (Zaretsky et al, 2016). Notably, the list of genomic alterations demonstrated to modify response to immune checkpoint blockade grows on a daily basis.
In addition to genomic alterations, epigenomic alterations in tumour cells may also have a profound effect on anti-tumour immune responses. Epigenetic chromatin modifications function physiologically to silence (or activate) genes in an orchestrated fashion during key developmental processes, however aberrant epigenomic alterations often exist in cancer, and can contribute to oncogenesis and also to immune evasion (Jones and Baylin, 2007). Epigenetic downregulation of antigen expression and silencing of immune-related genes may negatively impact immunotherapy response (Heninger et al, 2015), and early studies combining epigenetic modifiers, such as hypomethylating agents and histone deacetylase inhibitors, with immune checkpoint inhibitors have shown promising results (Wrangle et al, 2013).
Tumour microenvironment
The most extensively discussed component of the tumour microenvironment (TME) other than cancer cells themselves is tumour infiltrating lymphocytes (TIL). The presence of TIL has long been known to confer a favourable prognosis (Galon et al, 2006), and a greater appreciation of the complexity of immune infiltrates with regard to phenotype and distribution of the infiltrating leukocytes is mounting. Traditional metrics of TIL density and distribution (e.g., central vs peripheral) and gross enumeration of the T-cell infiltrate by CD3 and CD8 markers can now be readily supplemented with detailed characterisation of numerous surface markers, expression of immunomodulatory molecules, and quantification of T-cell clonotypes. Studies incorporating these techniques have revealed a broad range of infiltrating lymphocytes far beyond the dichotomous effector and regulatory T lymphocyte subsets, and have highlighted their complex regulatory potential as well as potential plasticity (Iida et al, 2011; Djenidi et al, 2015). Additional information has been gained by studying spatial relationships of TIL to tumour and stromal cells, yielding insight into the physical limitations to intercellular functional interactions. This has been demonstrated in the context of response to PD-1 blockade, in which not only density of CD8 T-cell infiltrate, but also location at the invasive margin and proximity of PD-1 expression to PD-L1 expression, were important factors associated with treatment response (Tumeh et al, 2014).
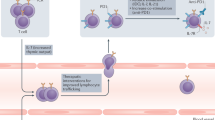
Tumours not only contain cancer cells, but also harbour a rich microenvironment composed of blood vessels, APCs, neutrophils, myeloid derived suppressor cells, tumour-associated macrophages and fibroblasts, components of the extracellular matrix, and soluble factors (such as cytokines and growth factors), all of which may assist or hinder anti-tumour immune responses. This is particularly evident when considering response to immune checkpoint inhibitors where the ability to exclude infiltrating immune cells from the TME can ‘make or break’ an anti-tumour immune response. On the basis of this, tumours have been classified into several cancer-immune phenotypes including ‘inflamed’ or ‘non-inflamed’ (Spranger and Gajewski, 2013), with more recent reports describing tumours as ‘immune-deserts’, ‘immune-excluded’, or ‘inflamed’ (Chen and Mellman, 2017). This type of classification motivates extensive research to identify predictive microenvironmental biomarkers that transcend existing markers such as PD-L1. Numerous therapeutic approaches targeting non-tumour cell stromal elements and functions are currently being tested in preclinical models and clinical trials, either as monotherapies or in combination with immune checkpoint blockade. Key examples include molecules inhibiting generation of the immunosuppressive metabolite indoleamine-2,3-dioxygenase (e.g., NCT02471846 and NCT 02073123), antagonists of the tumour-associated macrophage stimulating CSF1R (e.g., NCT02713529, NCT02526017, and NCT02323191), and ongoing development of agonist agents of the stimulator of interferon genes, aiming to favourably skew the TME towards an inflamed phenotype. Early studies of combination immune checkpoint and angiogenesis inhibition have showed promise from this multi-targeted approach in patients with advanced melanoma and renal cell cancer (Hodi et al, 2014; Wallin et al, 2016). Future advances in such strategies will be based on a deeper unravelling of the microenvironmental interactions to identify targetable nodes in the network.
Host immunity
Central to the efficacy of immune checkpoint blockade is preserved host immunity, predicated upon adequate number, availability, and activity of other innate and adaptive immune cell types. Systemic immunity is dynamic, shaped by prior antigenic stimuli, and influenced by interactions both within and outside the host, as diverse as invading microbial pathogens at topologically ‘external’ body surfaces (e.g., skin and gut), and interactions with tumours themselves. During the development and progression of cancer, components of dying tumour cells are taken up by APCs, which present processed antigen in the context of HLA to helper (CD4+) and cytotoxic (CD8+) T lymphocytes. This results in a cascade of events that leads ultimately to activation and expansion or anergy, depending on numerous modulating factors, principally the availability of appropriate co-stimulatory – or inhibitory – ligand-receptor engagement. This forms the foundation of the cancer immunity cycle described by Chen and Mellman (2013), and involves contributions from numerous other cells of the innate (e.g., NK cells) and adaptive (e.g., B lymphocytes) immune system. That a quantitatively and qualitatively intact overall immune system is important in cancer control is clear from the generally higher rates of many cancer types, including several without known viral aetiology, in immunosuppressed patients (Grulich et al, 2007). Although germline polymorphisms in immune-related genes are known to impact cancer predisposition and immune function in other settings such as haematopoietic transplantation outcomes (Delgado et al, 2010), whether polymorphisms predictive of checkpoint inhibitor efficacy or toxicity will be identified is currently unknown (though is highly likely).
The host T-cell repertoire, within which a subset of potentially tumour-reactive T cells resides, is largely shaped during development and early childhood while other components of the immune system remain more malleable throughout adult life. Host immunity moulds the tumour landscape, exemplified by the concept of immune editing as described by Schreiber and colleagues, through which immune action shapes the tumour composition to arrive at the parallel fates of equilibrium, elimination, or escape (Schreiber et al, 2011). However, it is becoming increasingly clear that this relationship is bidirectional; tumours may themselves profoundly influence the systemic environment through secretion of immunosuppressive cytokines and tumour-associated exosomes, which have been shown to be immunosuppressive (Meehan and Vella, 2016) and prime secondary locations for future metastasis (Peinado et al, 2012).
Environment
Importantly, factors within the broader environment (both outside and inside the host) may shape anti-tumour and overall immune responses. Perhaps the most poignant example of this is the microbiome, with recent evidence demonstrating a critical link between the gut microbiome and anti-tumour immunity. These interactions have significant implications in the setting of immune checkpoint blockade, as there is evidence that modulating the gut microbiome can enhance – or may even be a prerequisite for – therapeutic responses to these agents in preclinical models (Sivan et al, 2015; Vetizou et al, 2015). This has recently been studied in patients, with data suggesting that differential bacterial ‘signatures’ exist in responders vs non-responders to immune checkpoint blockade (namely, anti-PD-1 therapy) in a cohort of patients with metastatic melanoma (Gopalakrishnan et al, 2017). This finding needs to be validated in larger cohorts and across cancer types, but provides formative evidence regarding the influence of environmental factors on tumour and host immunity.
Indeed, other external pressures, such as diet and stress, can also impact the host and anti-tumour immunity (Kokolus et al, 2013), with hints that these factors might also modulate responses to immune checkpoint blockade although the complex mechanisms behind these influences are still being elucidated. Nonetheless, it is clear that we are gaining a more holistic and comprehensive view of the influences on anti-tumour immunity, which tightly relate to our understanding of the factors affecting therapeutic immune checkpoint blockade.
Implications for immune monitoring and novel strategies to overcome resistance to immune checkpoint blockade
On the basis of a deeper understanding of these pillars and hallmarks and the complex interactions between them, we will ultimately be able to refine strategies to monitor and enhance responses to immune checkpoint blockade. Importantly, the insights gained from the study of checkpoint inhibitor agents in current clinical use will have direct relevance to other forms of immunotherapy in active development, such as immunostimulatory checkpoint agonists and adoptive cell therapy.
The pillars (and hallmarks) of response to immune checkpoint blockade should be considered when designing immune monitoring strategies for these forms of therapy, and must take into account aspects of the tumour, the TME, overall immune competence, and environmental influences. This is already being done in some regards, with interrogation of specific genomic alterations and total mutational load as well as examination of the tumour for CD8 infiltrate density and PD-L1 expression. Evidence is emerging that monitoring host immune responses (e.g., via phenotypic markers, such as ICOS on T cells) (Ng Tang et al, 2013), may help predict responses to immune checkpoint blockade, and that the microbiome may serve as a predictive factor for long-term benefit to other forms of immunotherapy (Taur et al, 2014). Standardised approaches for each of these strategies are not yet developed and represent an area of unmet need in the field; additional intricacies will undoubtedly arise when monitoring combination therapies pairing immune checkpoint blockade with immunostimulatory agents (e.g., agonistic antibodies targeting 4-1BB or OX40), cell-based therapies, or molecular-targeted agents.
In addition to implications for monitoring responses, an understanding of these pillars and hallmarks also provides a framework for understanding and overcoming mechanisms of therapeutic resistance to immune checkpoint inhibitors. Numerous (epi)genomic, microenvironmental, and immune mechanisms of resistance to immune checkpoint blockade have been identified (Sharma et al, 2017), spurring the development of even more numerous multi-drug strategies targeting them. There is growing interest in better understanding the role of chronic inflammation, diet, and stress on general and tumour-specific immunity but much work is required to extract actionable elements from this knowledge.
The proposed pillars and hallmarks provide a foundation on which to build as we gain volumes of information from preclinical studies, clinical trials, and biomarker assessment in patients on standard-of-care therapy. Ultimately, integration of such data sets will inform optimal therapeutic strategies incorporating immune checkpoint blockade (and other forms of immunotherapy) in this age of cancer precision medicine.
References
Blankenstein T, Coulie PG, Gilboa E, Jaffee EM (2012) The determinants of tumour immunogenicity. Nat Rev Cancer 12 (4): 307–313.
Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE, Tsao H, Wargo JA (2010) Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res 70 (13): 5213–5219.
Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39 (1): 1–10.
Chen DS, Mellman I (2017) Elements of cancer immunity and the cancer-immune set point. Nature 541 (7637): 321–330.
Delgado DC, Hank JA, Kolesar J, Lorentzen D, Gan J, Seo S, Kim K, Shusterman S, Gillies SD, Reisfeld RA, Yang R, Gadbaw B, DeSantes KB, London WB, Seeger RC, Maris JM, Sondel PM (2010) Genotypes of NK cell KIR receptors, their ligands, and Fcgamma receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res 70 (23): 9554–9561.
Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpreville V, Validire P, Besse B, Mami-Chouaib F (2015) CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol 194 (7): 3475–3486.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313 (5795): 1960–1964.
Gopalakrishnan V, Spencer CS, Reuben A, Karpinets T, Hutchinson D, Hoffman K, Prieto PA, Tetzlaff MT, Lazar A, Davies MA, Gershenwald JE, Jenq RR, Hwu P, Sharma P, Allison JP, Futreal A, Ajami N, Petrosino J, Daniel-MacDougall C, Wargo JA (2017) Association of diversity and composition of the gut microbiome with differential responses to PD-1 based therapy in patients with metastatic melanoma. 2017 ASCO-SITC Clinical Immuno-Oncology Symposium; Orlando, FL, USA. J Clin Oncol 35(suppl 7S): abstract 2.
Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM (2007) Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 370 (9581): 59–67.
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5): 646–674.
Heninger E, Krueger TE, Lang JM (2015) Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol 6: 29.
Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, Zeng W, Giobbie-Hurder A, Atkins MB, Ibrahim N, Friedlander P, Flaherty KT, Murphy GF, Rodig S, Velazquez EF, Mihm MC Jr, Russell S, DiPiro PJ, Yap JT, Ramaiya N, Van den Abbeele AD, Gargano M, McDermott D (2014) Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res 2 (7): 632–642.
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363 (8): 711–723.
Iida T, Iwahashi M, Katsuda M, Ishida K, Nakamori M, Nakamura M, Naka T, Ojima T, Ueda K, Hayata K, Nakamura Y, Yamaue H (2011) Tumor-infiltrating CD4+ Th17 cells produce IL-17 in tumor microenvironment and promote tumor progression in human gastric cancer. Oncol Rep 25 (5): 1271–1277.
Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128 (4): 683–692.
Kokolus KM, Capitano ML, Lee CT, Eng JW, Waight JD, Hylander BL, Sexton S, Hong CC, Gordon CJ, Abrams SI, Repasky EA (2013) Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc Natl Acad Sci USA 110 (50): 20176–20181.
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, Palakurthi S, Fecci PE, Johnson BE, Janne PA, Engelman JA, Gangadharan SP, Costa DB, Freeman GJ, Bueno R, Hodi FS, Dranoff G, Wong KK, Hammerman PS (2016) Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 7: 10501.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373 (1): 23–34.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr (2015) PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372 (26): 2509–2520.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, Mitter R, Akarca AU, Linares J, Marafioti T, Henry JY, Van Allen EM, Miao D, Schilling B, Schadendorf D, Garraway LA, Makarov V, Rizvi NA, Snyder A, Hellmann MD, Merghoub T, Wolchok JD, Shukla SA, Wu CJ, Peggs KS, Chan TA, Hadrup SR, Quezada SA, Swanton C (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351 (6280): 1463–1469.
Meehan K, Vella LJ (2016) The contribution of tumour-derived exosomes to the hallmarks of cancer. Crit Rev Clin Lab Sci 53 (2): 121–131.
Ng Tang D, Shen Y, Sun J, Wen S, Wolchok JD, Yuan J, Allison JP, Sharma P (2013) Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol Res 1 (4): 229–234.
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12 (4): 252–264.
Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D (2012) Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18 (6): 883–891.
Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, Williams LJ, Deng W, Chen G, Mbofung R, Lazar AJ, Torres-Cabala CA, Cooper ZA, Chen PL, Tieu TN, Spranger S, Yu X, Bernatchez C, Forget MA, Haymaker C, Amaria R, McQuade JL, Glitza IC, Cascone T, Li HS, Kwong LN, Heffernan TP, Hu J, Bassett RL Jr, Bosenberg MW, Woodman SE, Overwijk WW, Lizee G, Roszik J, Gajewski TF, Wargo JA, Gershenwald JE, Radvanyi L, Davies MA, Hwu P (2016) Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 6 (2): 202–216.
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, Salama AK, Taylor M, Ott PA, Rollin LM, Horak C, Gagnier P, Wolchok JD, Hodi FS (2015) Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372 (21): 2006–2017.
Ribas A, Robert C, Hodi FS, Wolchok JD, Joshua AM, Hwu WJ, Weber JS, Zarour HM, Kefford R, Loboda A, Albright A, Kang SP, Ebbinghaus S, Yearley J, Murphy E, Nebozhyn M, Lunceford JK, McClanahan T, Ayers M, Daud A (2015) Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. 2015 ASCO Annual Meeting; Chicago, IL, USA. J Clin Oncol 33(suppl): abstr 3001.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348 (6230): 124–128.
Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331 (6024): 1565–1570.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A (2017) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168 (4): 707–723.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, Chang EB, Gajewski TF (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350 (6264): 1084–1089.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371 (23): 2189–2199.
Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523 (7559): 231–235.
Spranger S, Gajewski T (2013) Rational combinations of immunotherapeutics that target discrete pathways. J Immunother Cancer 1: 16.
Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF (2013) Up-regulation of PD-L1, IDO, and T (regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 5 (200): 200ra116.
Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, Gajewski TF (2016) Molecular drivers of the non-T-cell-inflamed tumor microenvironment in urothelial bladder cancer. Cancer Immunol Res 4 (7): 563–568.
Taur Y, Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, No D, Gobourne A, Viale A, Dahi PB, Ponce DM, Barker JN, Giralt S, van den Brink M, Pamer EG (2014) The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 124 (7): 1174–1182.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515 (7528): 568–571.
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D, Garraway LA (2015) Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350 (6257): 207–211.
Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, Poirier-Colame V, Roux A, Becharef S, Formenti S, Golden E, Cording S, Eberl G, Schlitzer A, Ginhoux F, Mani S, Yamazaki T, Jacquelot N, Enot DP, Berard M, Nigou J, Opolon P, Eggermont A, Woerther PL, Chachaty E, Chaput N, Robert C, Mateus C, Kroemer G, Raoult D, Boneca IG, Carbonnel F, Chamaillard M, Zitvogel L (2015) Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350 (6264): 1079–1084.
Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, Hernandez G, Mier J, He X, Hodi FS, Denker M, Leveque V, Canamero M, Babitski G, Koeppen H, Ziai J, Sharma N, Gaire F, Chen DS, Waterkamp D, Hegde PS, McDermott DF (2016) Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun 7: 12624.
Wherry EJ, Kurachi M (2015) Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15 (8): 486–499.
Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, Vancriekinge W, Demeyer T, Du Z, Parsana P, Rodgers K, Yen RW, Zahnow CA, Taube JM, Brahmer JR, Tykodi SS, Easton K, Carvajal RD, Jones PA, Laird PW, Weisenberger DJ, Tsai S, Juergens RA, Topalian SL, Rudin CM, Brock MV, Pardoll D, Baylin SB (2013) Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 4 (11): 2067–2079.
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TN, Lo RS, Ribas A (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375 (9): 819–829.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
JAW has honoraria from speakers’ bureau of Dava Oncology, Bristol-Myers Squibb, and Illumina and is an advisory board member for GlaxoSmithKline, Novartis, and Roche/Genentech. APC and MCA report no relevant conflict of interest or financial disclosures.
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Cogdill, A., Andrews, M. & Wargo, J. Hallmarks of response to immune checkpoint blockade. Br J Cancer 117, 1–7 (2017). https://doi.org/10.1038/bjc.2017.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.136
Keywords
This article is cited by
-
Targeting ubiquitin specific proteases (USPs) in cancer immunotherapy: from basic research to preclinical application
Journal of Experimental & Clinical Cancer Research (2023)
-
Exploring the role of pyroptosis in shaping the tumor microenvironment of colorectal cancer by bulk and single-cell RNA sequencing
Cancer Cell International (2023)
-
Radiotherapy remodels the tumor microenvironment for enhancing immunotherapeutic sensitivity
Cell Death & Disease (2023)
-
Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed
Biomarker Research (2022)
-
circCsnk1g3- and circAnkib1-regulated interferon responses in sarcoma promote tumorigenesis by shaping the immune microenvironment
Nature Communications (2022)
