
Abstract
Cytokines are involved in directing the activation of natural killer (NK) cells. NK cells are involved in the recognition of cells that have been altered; thus they do not recognize specific insults to the host, but when activated, are capable of destroying infected cells directly, as well as promoting the recruitment and response of the other components of the immune system by the release of cytokines and chemokines. It is these properties that have made NK cells a critical part of innate immunity and adaptive immunity, and they play a principal role linking innate and adaptive immunity by the recruitment of an adaptive immune response to an innate immune reaction.
Similar content being viewed by others
Introduction
NK cells are critical components of the host innate immune system in that they kill viral and other pathogen infected cells as well as provide surveillance against the development of malignancies 1, 2, 3, 4, 5. The activity and biological response of natural killer (NK) cells is controlled by inhibitory and activating cell surface receptors. These receptors may be triggered by multiple types of ligands including membrane bound molecules such as MHC class 1 or the Rae family of proteins 1, 2, 3, 4, 5 as well soluble mediators released by other cells that both activate and inhibit NK function (e.g., Activating-IL-2, IL-12, IL-15, IL-18, IFN-α; Inhibiting-IL-10, TGF-β) 1, 2. The inhibitory receptors serve to moderate NK activity by dampening cytokine release and cytotoxicity if their cognate ligands are triggered by interaction with their receptors on the NK cells. This dampening effect is critical in order to prevent wide scale destruction of the host by the NK component of the innate immune system. Our current studies have focused on the role of cytokine signaling in the context of an environment where both NK activating and inhibitory receptors are triggered. Our findings will be summarized here.
The model
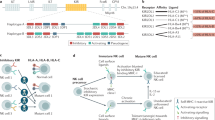
In the context of the immune response, numerous factors play a role in dictating the response of any specific cell type to the local environment. With respect to NK cells, the NK target cell interaction is likely to involve crosslinking of activating and inhibitory receptors on the cell surface. Cross linking of activating receptors results in a triggering of gene activation with the release of both cytokines and chemokines (Figure 1).

Crosslinking of activating receptors results in cytokine/chemokine gene expression.
The simultaneous crosslinking of both activating and inhibitory receptors on the NK cell (Figure 2) results in a blockage of NK activity as NK cytokine release is suppressed and activation of the cytotoxic pathways (e.g., perforin, Fas, TRAIL) is dampened. However the NK cell-target cell interaction may occur in the presence of a macrophage or other monocytic cell (e.g., dendritic cell) that has been infected by a virus, bacteria or other pathogen (Figure 3).

Crosslinking of both activating and inhibitory receptors leads to inhibition of the NK response.

Pathogen infection of a macrophage or dendritic cell can lead to release of soluble NK cell activators.
The release of soluble immune mediators (e.g., IL-12, IL-18, IFNγ, etc) from the infected cells can then feed back onto the NK cells (Figure 4).

Soluble mediators released by activated macrophages or dendritic cells feed back on NK cells to enhance IFNγ release.
This feedback results in a synergistic response with respect to the NK cell expression of Interferon gamma (IFNγ) (Figure 4). In addition other cytokines and chemokines are released to further attract other immune effector cells to the site of inflammation. Thus the combination of cell surface receptor activation with soluble mediators released from other cells initiates the cytokine cascade with IFNg being the major product. This cytokine release then triggers a strong host response.
Results
Previous studies from our laboratory have identified the effects on NK cells of crosslinking LY49 activating receptors 6. These activating receptors, which bind MHC class I ligands (e.g., H2-Dd), contain cytoplasmic tyrosine based activating motifs and depend upon an accessory molecule, DAP 12, for their function 7, 8. In these and other studies 9, 10 we demonstrated that cross-linking of the LY49D activating receptor resulted in the release of a range of cytokines and chemokines from NK cells, thus indicating that a major function of the activating receptors was triggering expression of these soluble immune mediators. Other laboratories have demonstrated that triggering of these receptors results in calcium mobilization and these receptors can mediate antibody dependent cellular cytotoxicity in the presence of specific antibodies 7, 8. We then determined that, upon addition of the cytokines IL-12 or IL-18, IFNγ but not other cytokines or chemokines was induced in a synergistic manner 9, 10. For example, as previously reported 9, treatment of 1 × 106/ml LY49D + liver NK cells with IL-12 for 6 h resulted in < 100 pg/ml of IFNγ, cross-linking of the LY49D receptor resulted in the production of close to 900 pg/ml of IFNγ but combining both signals resulted in > 7700 pg/ml of IFNγ. Furthermore, as we recently reported 11, other IL-12 family members, IL-23 and IL-27, also demonstrated a similar, but weaker, co-activation signal with ITAM bearing receptors, and the IL-12 synergy was STAT4 dependent, as no enhanced IFNg expression was observed when cells from STAT4 KO mice were treated in the presence or absence of IL-12 11. This synergism was observed after the cells were pretreated with IL-12 for as little as 15 min -30 min and the cells remained hyperresponsive to LY49 cross linking for up to 3 h after the IL-12 treatment 11. We have extended these studies to analyze the regulation of other ITAM positive receptor signaling on NK, NKT and T cells. We have found that crosslinking of NKG2D and NK1.1 on NK cells results in a synergistic NK IFNg response when combined with IL-12 or IL-18 11. Furthermore cross-linking of NKG2D and CD3 on NKT and T cells resulted in potent synergy with respect to IFNg expression but not TNFa expression when combined with IL-12, and to a lesser degree with IL-18. The CD3 + IL-12 synergy in T cells confirms data previously reported by other investigators 12. Interestingly, crosslinking the NKT cells with a GalCer also resulted in a synergistic induction of IFNg when combined with IL-12 11. Mechanistic examination of the synergy indicated a potent upregulation of total IFNg mRNA in both the nuclear and cytoplasmic compartment without affecting mRNA halflife 11. Interestingly the overall synergy was further enhanced by combinations of sub-optimal doses of IL-12 and IL-18 with both Ly49s and NKG2D (in vitro and in vivo 11) thus indicating the physiological relevance of our findings.
Next we investigated the biochemical pathways involved in the induction of IFNg gene expression. Upon flow cytometric analysis we determined that IL-12 treatment resulted in prolonged STAT4 phosphorylation and p38 MAP kinase phosphorylation 11, consistent with what has been previously reported by many laboratories. Furthermore and again as expected, LY49D cross linking resulted in increased phosphoErk. By using pharmacological inhibitors of these pathways (SB203580 for p38 and U0126 for Erk), we completely blocked the synergistic expression of IFNg 11, thus demonstrating that both pathways are critical for the synergistic increase in gene expression. These results are most consistent with the model proposed by Nakahira and colleagues 13 where increased pSTAT4 (resulting from the IL-12 treatment) results in increased recruitment of AP-1 to the IFNg promoter [note that in our model, cross-linking LY49D results in increased AP-1 activity, (McVicar DW personal communication)]
To extend these studies, we next asked whether or not cytokine signaling could overcome the effects of inhibitory receptors. These inhibitory receptors are critical to NK function as they prevent the NK cells from killing inappropriate targets. We had previously shown that if activating and inhibitory receptors are triggered simultaneously through NK-target cell interactions, the production of IFNg by NK cells is inhibited by at least 60% 9. Thus the inhibitory signal acts to suppress a strong response by the NK cells and thus temper the initial host response to a potential insult. We found that when adding either IL-12 or IL-18 in the presence of both activating (LY49D) and inhibitory receptors (LY49I), the activation signals dominated and IFNg was expressed at levels equivalent to that observed with the activating receptor and cytokine alone 9, 11. This synergy was also seen at the RNA level 11. As the inhibitory receptors can also attenuate the NK cell response of crosslinking other activating receptors on the cell surface (NKG2D, NK1.1, LY49H), we next investigated whether or not cytokines would be the key second signal in overcoming the dominant effects of inhibitory receptor cross-linking. Our results 11 confirmed the potent effects of IL-12, as synergistic activation of IFNγ gene expression was observed when any of the activating receptors were crosslinked in the presence of the LY49I inhibitory receptor. Thus, as proposed in the model depicted above, cytokines, as soluble mediators of the immune response, represent a signal critical to maximizing the host response to insult.
Summary and conclusions
Our data demonstrates that multiple convergent signals can maximize the innate immune response of NK cells to potential danger signals. As an integral part of the host innate immune response, activation of NK cytokine expression and antibody dependent complement mediated cytotoxicity (ADCC) killing ability takes place when an LY49 activating receptor is triggered through interaction of the receptor with the appropriate MHC class I surface antigen on a target cell. These are important functions of the NK cells, as a major role for these cells is to eliminate cells from the host that have become damaged or altered as a result of invasion by a pathogen or malignant transformation. As NK cells are normally kept quiescent with respect to cytokine gene expression, cross-linking of the activating receptor is sufficient to result in some cytokine gene expression. However as we have demonstrated, the response is maximized when second signals are received from other leukocyte populations (e.g., macrophages or dendritic cells). The presence of these second signals (IL-12, IL-18, Interferon-α) are a direct result of pattern recognition receptor (i.e., Toll receptor) activation by viruses, bacteria, fungi or other tissue insult. Thus the immune system begins to “ramp up” in order to maximize the innate immune response when multiple cytokines are released into the localized microenvironment. The impact of the soluble mediators is critical for overcoming the function of the inhibitory receptors on the NK cells as these receptors are programmed to moderate the overall NK response. Most interestingly, IFNg appears to be the gene most sensitive to multiple signals as expression of IFNγ, but not that of other cytokines, is synergistically increased when cell surface receptors are cross-linked in the presence of cytokines. This increased IFNγ is essential for activating macrophages in the area of insult as well as increasing MHC class I and MHC class II cell surface expression on potential target cells, thus enhancing the ability of the adaptive immune system to respond to challenge. Both the p38 MAPK and Erk pathways appear to be involved in the increased IFNγ expression and these pathways activate multiple transcription factors required for IFNγ transcription, including STAT4 and AP-1.
Another consideration is the potential role for NK cells in the generation of the adaptive arms of immunity. As elucidated in a recent review regarding autoimmunity 14, future studies will probably show a similarly important role for innate cells, including NK cells, in these and other adaptive responses. It is clear that NK cells can provide signals to both DC and T cells through soluble factors secreted by the NK cells or direct cell-cell interactions and that these signals trigger effector functions in the responder cells. Thus the role of NK cells and the consequence of the local microenvironment in which they exist and function are becoming critical components in understanding the initial host immune response. Therefore an important issue for the future will be the dissection of the heterogeneity in NK- target cell responses and the relationship of these responses to NK-cell subsets, the local cytokine environment and the expression pattern of activating and inhibitory NK cell surface receptors. Thus NK cells and their response to stimuli are becoming a central component in regulating both the initial innate immune response and subsequent adaptive immune response of the host. Developing methods to control and appropriately maximizing this response may offer new therapeutic approaches to controlling tumor development and the ability of pathogens to spread within the host.
References
Blach-Olszewska Z . Innate immunity: cells, receptors, and signaling pathways. Arch Immunol Ther Exp (Warsz) 2005; 53:245–53.
Bottino C, Castriconi R, Moretta L, Moretta A . Cellular ligands of activating NK receptors. Trends Immunol 2005; 26:221–6.
Wallace ME, Smyth MJ . The role of natural killer cells in tumor control—effectors and regulators of adaptive immunity. Springer Semin Immunopathol 2005; 27:49–64.
Dimasi N, Biassoni R . Structural and functional aspects of the Ly49 natural killer cell receptors. Immunol Cell Biol 2005; 83:1–8.
Hamerman JA, Ogasawara K, Lanier LL . NK cells in innate immunity. Curr Opin Immunol 2005; 17:29–35.
Ortaldo JR, Bere EW, Hodge D, Young HA . Activating Ly-49 NK receptors: central role in cytokine and chemokine production. J Immunol 2001; 166:4994–9.
Mason LH, Willette-Brown J, Anderson SK, et al. Characterization of an associated 16-kDa tyrosine phosphoprotein required for Ly-49D signal transduction. J Immunol 1998; 160:4148–52.
Smith KA, Wu J, Bakker ABH, Phillips JH, Lanier LL . Ly49D and Ly49H associate with mouse DAP12 and form activating receptors. J Immunol 1998; 161:7–10.
Ortaldo JR, Young HA . Mouse Ly49 NK receptors: balancing activation and inhibition. Mol Immunol 2005; 42:445–50.
Ortaldo JR, Young HA . Expression of IFN-gamma upon triggering of activating Ly49D NK receptors in vitro and in vivo: costimulation with IL-12 or IL-18 overrides inhibitory receptors. J Immunol 2003; 170:1763–9.
Ortaldo JR, Winkler-Pickett R, Wigginton J, et al. Regulation of ITAM positive receptors: role of IL-12 and IL-18. Blood 2005; Oct 25, [Epub ahead of print]
Leite-de-Moraes MC, Moreau G, Arnould A, et al. IL-4-producing NK T cells are biased towards IFN-gamma production by IL-12. Influence of the microenvironment on the functional capacities of NK T cells. Eur J Immunol 1998; 28:1507–15.
Nakahira M, Ahn HJ, Park WR, et al. Synergy of IL-12 and IL-18 for IFN-gamma Gene Expression: IL-12-Induced STAT4 Contributes to IFN-gamma Promoter Activation by Up-Regulating the Binding Activity of IL-18-Induced Activator Protein 1. J Immunol 2002; 168:1146–53.
Johansson S, Berg L, Hall H, Hoglund P . NK cells: elusive players in autoimmunity. Trends Immunol 2005; 26:613–8.
Acknowledgements
We wish to acknowledge the work of Dr Deborah Hodge, Dr Dan McVicar, Mike Sanford, Della Reynolds, Anna Mason, Bill Bere, John Wine and Robin Winkler-Pickett that contributed to this project. This project was supported by funding from the Intramural Research Program, Center for Cancer Research, National Cancer Institute-Frederick, National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Young, H., Ortaldo, J. Cytokines as critical co-stimulatory molecules in modulating the immune response of natural killer cells. Cell Res 16, 20–24 (2006). https://doi.org/10.1038/sj.cr.7310004
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310004
Keywords
This article is cited by
-
Microwave ablation of primary breast cancer inhibits metastatic progression in model mice via activation of natural killer cells
Cellular & Molecular Immunology (2021)
-
NK cells mediate the cumulative analgesic effect of electroacupuncture in a rat model of neuropathic pain
BMC Complementary and Alternative Medicine (2014)
-
G-CSF downregulates natural killer cell-mediated cytotoxicity in donors for hematopoietic SCT
Bone Marrow Transplantation (2012)
