
Abstract
Based on ecological and behavioural studies it has been assumed that Eurasian perch (Perca fluviatilis) within one lake may not represent one panmictic population, but that they are subdivided into subpopulations. In order to investigate the genetic substructuring of populations, we used gene frequencies of five microsatellite loci to compare perch from six different sites from Lake Constance, Germany, and as outgroups perch from the lake Grosser Vätersee, Berlin, and two Swiss lakes, Lake Zurich and Lake Walensee. We examined whether homing behaviour of subadults to the spawning sites of their parents occurs and whether philopatric behaviour of adults results in significant population genetic substructuring. The distribution of genetic variation revealed two major, genetically distinct populations in Lake Constance: one in the eastern part of the lake and another in the western part (GST=0.07). Within each of these two populations, no further genetic substructuring, nor any indication of inbreeding could be detected, either because genetic exchange was sufficiently high or because the time since separation has been too short. Homing behaviour of subadults to parental spawning sites after having spent several weeks of their life cycle in the pelagic zone could not be detected. Instead, subadults stay within either the western or the eastern region of the lake. There is evidence that some shoals contain full- and half-sibs. Despite females spawning in close proximity to each other, some siblings stay together. This might suggest that perch possess kin preferences and kin recognition.
Similar content being viewed by others


Introduction
Recently, analyses of genetic population structure in several aquatic species have generally found that individuals from different parts of a lake or tributary do not form a single panmictic population, but instead are typically genetically substructured into different local populations (Carvalho, 1993; Tessier et al., 1997; Brunner et al., 1998; Chenoweth et al., 1998). It has been assumed that the number of these subpopulations is a function of the number of geographical or physical barriers encountered by a species during its life cycle (Sinclair, 1988). Genetic drift or differential selection regimes in these isolated local populations might play an important role in the evolution of new species (Dieckmann & Doebeli, 1999; Kondrashov & Kondrashov, 1999). Genetic isolation of local populations is caused, among other reasons, by philopatric behaviour and local mating of adults and homing behaviour of subadults which return to parental spawning sites after spending a part of their life cycle in different habitats.
Female perch over three years old spawn from April to June, attaching their eggs in up to 1 m long strands on stones and other structures in the littoral zone. After hatching, larvae stay for approximately two weeks in this area before they drift into the pelagic zone where they live for about one month (Wang & Eckmann, 1994). In July, after the abundance of pelagic zooplankton decreases, juvenile perch return to the shallow littoral zones. If homing behaviour existed, then individuals from a specific locality would be expected to be genetically more similar to each other and to show significant genetic difference from those of other localities.
To date, behavioural and ecological studies have led to the assumption of sedentary behaviour in adult Eurasian perch (Perca fluviatilis). For example, in Lake IJssel in the Netherlands, Willemsen (1977) marked and released adult Perca fluviatilis away from their capture site; later, he recaptured them at the place of first capture. Similar results were reported for perch from the English Lake Windermere (Kipling & Le Cren, 1984). Analysis of allozyme variation revealed the existence of two distinct populations in the northern and southern parts of Lake Windermere (Bodaly et al., 1989). Perch from Lake Constance also carry different intensities of parasite infection according to their capture sites, suggesting that adult fish are resident, possibly philopatric, and have little contact with populations from other localities in the lake (Balling, 1992; Dieterich, 1998).
Homing behaviour spectacularly demonstrates the often astonishing ability of a wide range of animals for orientation and navigation. Atlantic and Pacific species of salmon return to their natal rivers after years at sea from a distance of hundreds or even thousands of kilometres (Hasler & Scholz, 1983; Gyllensten & Wilson, 1987). Similarly, Oceanic loggerhead turtles (Caretta caretta) and green turtles (Chelonia mydas) return to spawn at the beach where they hatched (Allard et al., 1994; Bowen et al., 1994; Bolten et al., 1998), and many bird species return to their nesting places after crossing whole continents (Dingle, 1996). Breeding in similar environmental conditions as the parental generation did can increase reproductive success and therefore favours selection for homing behaviour. A DNA microsatellite analysis was undertaken to examine the genetic diversity of perch populations of different ages from several localities in the littoral zone of Lake Constance and from three other lakes in Germany and Switzerland to evaluate genetic substructuring and homing behaviour.
Materials and methods
Sampling
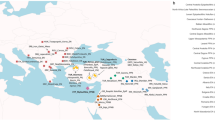
Perch were collected at six different localities in Lake Constance (see Fig. 1) during the winter of 1997/1998. Additionally, perch from three other lakes were included in this study: Grosser Vätersee, near Berlin, Germany, and Lake Zurich and Lake Walensee, Switzerland. At locality 5, animals were sampled by electro-fishing along a reed belt over a distance of 400 m (Fig. 1). A trawl net was used to sample at locality 1; all other localities were sampled with bottom set gill nets. All animals per locality were of the same age; but the age of the perch differed among localities: 0 + (caught before one year): OG, KR, GN; 1 + (caught before 2 years of age): US, B; 2 + (caught before 3 years of age): LA, RO; 3 + (caught before 4 years of age): LW, LZ. DNA microsatellite analysis was performed on 18 individuals per locality.

Map of sampling localities of perch in Lake Constance: 1 OG Obere Güll, 2 LA Langenargen, 3 RO Romanshorn, 4 KR Kreuzlingen, 5 GN Gnadensee, 6 US Untersee, and in two lakes in Switzerland: 8 LW Lake Walensee and 9 LZ Lake Zurich. Arrows indicate the main current in Lake Constance.
Microsatellite analysis
Muscle and liver tissues were stored in 80% ethanol until DNA extraction. Genomic DNA was extracted according to standard phenol–chloroform procedures (Sambrook et al., 1989). Polymerase chain reaction (PCR) amplification and genotyping were conducted for five dinucleotide microsatellite loci (CA and GT repeats), using primers developed for walleye (Stizostedion vitreum) (Borer et al., 1999) (Table 1). Amplification products (1.8 μL) were electrophorized on 6% denaturing polyacrylamide gels for 3.5–6 h and visualized using X-ray-sensitive films. The allele sizes found for each locus were determined and numbered according to their length (e.g. allele 1 being the smallest).
Data analyses
Genetic diversity was quantified in terms of heterozygosity, number of alleles per locus and allele frequencies observed in all local populations. GENEPOP version 1.2 (Raymond & Rousset, 1995) was used for estimating deviations from Hardy–Weinberg equilibrium at each locus and globally over all loci. Using FSTAT version 2.8 (Goudet, 1995) FIS-values (Weir, 1990) and GST values were assessed by jackknifing procedures (1000 iterations) and used for calculations of relatedness within local populations. According to Wright (1969) who described genetic population structures in terms of the F-statistics, the relationship between FST, FIT and FIS can be written as (1 − FIT)=(1 − FST)(1 − FIS). It is possible to partition the intraclass relatedness ‘r’ in the same way (Pamilo, 1984). The relatedness values in a subdivided population can be expressed as (1 − rGT) = (1 − rST)(1 − rGS) where rGS is the average relatedness among individuals in social groups within subpopulations, rST is the average relatedness of individuals among subpopulations, and rGT is the average relatedness of individuals in groups with reference to the total populations (Pamilo, 1989). Therefore, intraclass relatedness, i.e. genotypic correlation among group members, can be written as rGS=2FGS/(1 + FIS), where FGS is the gametic correlation of group members within a subpopulation and FIS measures inbreeding. Pamilo (1984, 1985) further proposed an inbreeding-corrected relatedness, which is estimated as: Relatc=[r − 2Capf/(1 + Capf)]/[1−2Capf/(1 + Capf)] in the program FSTAT 2.8, where Capf is an equivalent to FIT. When the population is structured, Relatc adequately removes the increase in relatedness due to this structuring (see Chapuisat et al., 1997).
Although it is straightforward to estimate the average relatedness in social groups, estimating values for specific individuals with, e.g. microsatellite data is still problematic: On average, full-sibs share 50% of their alleles and half-sibs 25%, but it depends on the frequency of alleles within the population how informative sharing alleles at a single locus is. The frequency of each allele within the population has to be taken into account for classifying individuals as being related or not. Therefore, we did not use the value of sharing 50% of the alleles as the criterion but calculated the threshold for being full- or half-sibs according to the allele frequencies of perch in Lake Constance by extending a method described by Blouin et al. (1996). We randomly generated 1000 pairs of unrelated individuals and calculated the number of shared alleles using SAS version 6.12 (SAS Institute, 1986). To evaluate the number of shared alleles of full-sibs we randomly chose 1000 pairs of individuals, generated two offspring each and calculated the matches of alleles between them. We used a similar procedure to generate the mean number of matched alleles for half-sibs. Dividing by the number of loci gives Mxy, the average number of matches per locus between individuals x and y. Using a logistic regression analysis (SAS Institute, 1995) we calculated the probabilities for each Mxy to belong to either of the three different groups (unrelated, half- or full-sibs) analogous to a likelihood ratio principle. The threshold for being classified as unrelated was Mxy ≤ 0.5, as half-sibs 0.5 > Mxy < 0.7, and as full-sibs Mxy ≥ 0.7. In a subsequent analysis we calculated Mxy for each pair of individual perch per sampling site and determined the number of full-sibs and half-sibs according to these thresholds. We determined whether the observed number of full- and half-sibs at each locality was higher than would be expected from combinations of unrelated animals which were misclassified as being related. The analysis was performed by 1000 simulations of constructed matrices which were derived by creating 18 virtual individuals on the basis of the gene frequency of perch in Lake Constance and comparing their alleles pairwise as described above. The performed significance test based on the null hypothesis that there were no full- or half-sibs among the sampled individuals. For each locality it was calculated if the observed numbers of full- and half-sibs were significantly different from this distribution.
Pairwise FST-values (θ-values) (Weir, 1990) were determined based on the gene frequencies using an analysis of molecular variance (AMOVA) (Michalakis & Excoffier, 1996) available in the program ARLEQUIN version 1.1 (Schneider et al., 1997). Probability values of the pairwise FST-values were adjusted for multiple simultaneous table-wide tests using the sequential Bonferroni adjustments (Rice, 1989) to minimize type-I errors. Genetic distance values were analysed according to Reynolds et al. (1983) with D=− ln (FST) calculated with the program GENETIX (Belkhir et al., 1997). Mantel matrix correlations (Mantel, 1967) were used to compare geographical (straight-line distance between different localities) and genetic distance D within Lake Constance.
Results
Population substructuring
Comparing all perch populations from Lake Constance, a FST-value of 0.054 was calculated. Genetic subdivision between local populations was examined by calculating pairwise FST-values (Table 2). According to these data, perch sampled from six different localities in Lake Constance can be subdivided into two large populations. The first population is composed of individuals from the east (localities 1, 2 and 3) and the second in the western part includes specimens from localities 4, 5, and 6 (Fig. 1). The GST value (corrected) for all loci between these two populations is 0.07 (P < 0.001) indicating a moderate genetic substructuring. Whereas perch from localities 1 and 2 differed significantly from those from localities 4, 5, and 6 the genetic exchange between localities 3–4 and 3–6 was higher. FST-values between these localities were moderate (0.051 and 0.048, respectively), but after Bonferroni corrections these values proved not to be statistically significant.
Perch from both Swiss lakes, Lake Walensee and Lake Zurich, were genetically similar (FST=0.022, P < 0.30). Interestingly, perch from Lake Zurich and Lake Walensee were genetically more similar to animals from the eastern part of Lake Constance (localities 1, 2, and 3) (FST=0.054, P < 0.001) than to those from the western part (localities 4, 5, and 6) (FST=0.112, P < 0.001). As expected, the highest and most statistically significant degree of genetic subdivision was observed between perch from the lake near Berlin and all other localities (mean FST-values=0.256, P < 0.001). A phenogram (Fig. 2) illustrates the different genetic distances (D-values) among perch from all localities studied.

Phenogram of genetic distances constructed by UPGMA between all investigated populations of perch. *Indicates statistically significant differences between the branches.
When genetic distances of populations from Lake Constance were plotted against the logarithm of geographical distances between populations, a significant, positive correlation (Mantel test; R2=0.26; P < 0.001) was found, indicating an isolation by distance effect (Slatkin, 1993).
Genetic structure of local populations
The degree of allelic polymorphism at five microsatellite loci varied between four and 10 alleles per locus (Table 1). Some alleles were only found in perch from one locality or a specific lake (e.g. alleles 1 and 4 of locus Svi6 found only in Lake Constance; alleles 3 and 5 of locus Svi26 found in the Grosser Vätersee near Berlin).
FIS-values varied from 0.068 to −0.276 and showed a statistically significant excess of heterozygotes in six out of nine subpopulations, as illustrated in Table 3. On average, over all loci and local populations of Lake Constance FIS=−0.117. The mean degree of relatedness between individuals per locality was r=0.115 using the calculation of Queller & Goodnight (1989) and Relatc =0.674 (relatedness corrected) (Pamilo, 1984, 1989; Goudet, 1995), indicating that animals collected at each locality were closely related. In order to test for kin aggregations we calculated pairwise genetic relatedness between all individuals of each locality using a program based on the SAS routine IML version 6.12 (SAS Institute, 1986) according to Blouin et al. (1996). Based on the distribution of allele frequencies of the total population of Lake Constance, individuals were classified as unrelated when Mxy ≤ 0.5, as half-sibs when 0.5 < Mxy < 0.7 or as full-sibs when Mxy ≥ 0.7. In four out of six localities the number of individuals which shared more than 50% of the alleles (classified as full- and half-sibs) or 70% of the alleles (full-sibs) was significantly higher than in randomly generated groups of 18 unrelated individuals. Therefore, we concluded that perch indeed stayed in groups of kin. Only at localities 3 and 4 did the number of observed pairs of animals classified as full-sibs not exceed the expected values.
We found the same degree of relatedness in animals of different age, which suggests that related animals shoal together even with increasing age.
Discussion
Two major, genetically different populations of the Eurasian perch exist in Lake Constance: one in the eastern part of the lake including localities 1, 2, and 3 and a second one in the western part including localities 4, 5, and 6 (Fig. 2). Genetic differentiation was indicated by a statistically significant GST value of 0.07. These results correspond to those of Bodaly et al. (1989), who could distinguish between two distinct populations of perch in the northern and southern part of the English lake Windermere using allozyme analysis.
Within each of these two populations in Lake Constance, pairwise FST-values do not differ significantly. Because adult perch are supposed to be sedentary (Willemsen, 1977; Kipling & Le Cren, 1984), the degree of inferred genetic exchange within these populations might depend on distribution patterns of the larvae. According to this scenario, there might be separate areas in the eastern and western part in Lake Constance where larvae spend their pelagic phase before they return to the littoral zone. To which bank of the lake early juveniles return might depend on wind-driven transport of surface waters, but there is a clear separation between the two parts of the lake.
FST-values between localities 3 (RO) and 4 (KR) and between 4 (KR) and 5 (GN) and 6 (US) were not significantly different from 0. This result indicates genetic exchange between animals of these localities which can be explained by transportation, i.e. of larvae by the main water currents in Lake Constance. The Rhine river flows into the lake at the south-east, near locality 3 (RO) (Fig. 1). Near KR, where the Rhine flows out of the lake, there is a strong, continuous current towards US (Fig. 1).
In this study fish were collected during the winter. Therefore, we do not know yet whether individuals spend some time at different sites but return to their parental spawning sites during the mating period in early summer, resulting in an even stronger genetic substructuring among localities. This might be the case in walleye (Stizostedion vitreum), a fish quite closely related to perch with a similar ecology, where tagging studies indicated that animals moved readily between lakes during the year (Nepszy et al., 1991); but significant differences in genotypic frequencies among spawning sites were consistent with the hypothesis of natal homing to spawning sites for both sexes during the mating season (Stepien & Faber, 1998). Experimentally supported natal homing was also found in walleye in laboratory-reared released broodstock. This investigation found that natal homing in spawning walleye is governed by a genetically based response to environmental cues (Jennings et al., 1996). More genetic data of perch from different localities should be collected during the spawning time. This would provide further evidence whether perch migrate during some periods of their life cycle but spawn at the same sites as their parents.
Perch from both Swiss lakes (Lake Zurich and Lake Walensee) which are interconnected by the Linth channel (Fig. 1) are genetically similar. The observation supports the assumption that animals use the channel to migrate between the lakes. Interestingly, perch from these lakes were genetically more similar to perch trapped in the eastern part of Lake Constance (FST=0.054, P < 0.001) than animals of the eastern and western parts of Lake Constance are to each other (FST=0.07, P < 0.001). This finding supports geological studies that there was a connection between the Swiss lakes and the eastern part of Lake Constance approximately 15 000 years ago (Kiefer, 1972). It also indicates that the genetic divergence between the two populations within Lake Constance is quite strong.
Our study reveals for the first time that perch were staying in shoals of half-siblings or more closely related individuals, as indicated by a high Relatc = 0.674 (Pamilo, 1984, 1989) and a high mean r=0.115 using the analysis of Queller & Goodnight (1989). The appearance of kin aggregations was also strongly supported using an extended method according to Blouin et al. (1996) revealing full- and half-sibs per locality. Blouin et al. (1996) demonstrated the ability of microsatellites to discriminate unrelated individuals from full-sibs with at least 97% accuracy. Attaining this level of accuracy necessitates using a large number of microsatellite loci. However, similar robustness was shown by Piertney et al. (1999) using a limited number of microsatellite loci for calculating pairwise relatedness.
The occurrence of shoals of full- and half-sibs can be explained by the mating system, but there have been few observations of mating under natural conditions; first attempts of filming spawning perch showed that multiple paternity may occur: two different males were observed fertilizing eggs of one female (Treasure, 1981).
Perch live in age-structured shoals (Wang & Eckmann, 1994; Imbrock et al., 1996). The same level of relatedness was found in shoals of 0 + and those of 2+ years old fish, which suggests that related animals stayed together not only during the larval phase but continued to do so with increasing age. Several species of fish spend periods of their life cycles in kin aggregations. This result has been also observed in the Atlantic cod (Gadus morhua) (Herbinger et al., 1997), coho salmon (Oncorhynchus kisutch) (Quinn & Busack, 1985), and in freshwater species like cichlids (Sarotherodon melanotheron) (Pouyaud et al., 1999), common shiners (Notropis cornutus) (Ferguson & Noakes, 1981), and three-spine sticklebacks Gasterosteus aculeatus (FitzGerald & Morrissette, 1992). Cichlids (Sarotherodon melanotheron) originating from riverine environments appeared to be panmictic, but in samples from open waters such as lagoons, showed sibling aggregations (Pouyaud et al., 1999). Fontaine & Dodson (1999) did not reveal kin-biased distribution among juveniles of Atlantic salmon. But this study was performed in a large river which coincides with our findings that animals are less related in areas with high water currents (localities 3 and 4). These highly dispersive environmental conditions are less conducive to the establishment of kin-biased distribution.
Shoaling behaviour is proposed to be an altruistic trait that may have evolved through kin selection (Pitcher, 1986). Individuals might increase their inclusive fitness by providing the benefits of cooperative behaviour to related individuals (Hamilton, 1964). The mechanisms by which these kin groups develop and stay distinct from other such groups is unknown. A kin recognition system might permit larvae to distinguish between kin and non-kin which hatch in close proximity to them. They might use olfactory cues from the eggs, as has been shown in tadpoles, which learn to distinguish between kin and non-kin during their early life phase (Blaustein & Waldman, 1992). Alternatively, female perch might mark the substrate where the eggs are laid, as is assumed for Atlantic salmon (Salmo salar) which occupy fixed territories on the stream bed during the summer months (Kalleberg, 1958; Keenleyside & Yamamoto, 1962). The substrate within each territory is probably marked with odours that can be recognized by kin (Stabell, 1987). Active compounds produced in the liver and expelled via the intestinal tracts could mediate this recognition (Quinn & Hara, 1986; Stabell, 1987). Laboratory experiments on recognition of kin vs. non-kin groups of Atlantic Salmon (Salmo salar) (Brown & Brown, 1993, 1996) reveal the possible advantage of kin aggregations. Kin groups had fewer aggressive interactions, used a greater proportion of ‘threat’ behaviour as opposed to fighting, and had improved growth, especially in subordinates.
Kin recognition mechanisms play an important role in assortative mating (Hepper, 1986). By avoiding breeding with close relatives but choosing genetically similar mating partners adaptations to specific environmental conditions are maintained which might be the basis for processes of speciation (Kondrashov, 1998; van Oppen et al., 1998). Additional behavioural studies of perch might be able to identify the behavioural mechanisms of these processes.
References
Allard, M. W., Myamoto, M. M., Bjorndal, K. A. and Bolten, A. B. et al1994). Support for natal homing in Green turtles from mitochondrial DNA sequences. Copeia, 1: 34–41.
Balling, T. (1992). Saisonale und standortabhängige Verbreitung von Fischparasiten im Bodensee-Obersee und ihr Einfluß auf den Ernährungszustand der Fische. PhD Thesis, Eberhard-Karls-Universität, Tübingen.
Belkhir, K., Borsa, P., Goudet, J. and Chikhi, L. et al1997). GENETIX v. 3.0, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome et Populations, CNRS UPR 9060, Université Montpellier 2, Montpellier.
Blaustein, A. R. and Waldman, B. (1992). Kin recognition in anuran amphibians. Anim Behav, 44: 207–221.
Blouin, M. S., Parsons, M., Lacaille, V. and Lotz, S. (1996). Use of microsatellite loci to classify individuals by relatedness. Mol Ecol, 5: 393–401.
Bodaly, R. A., Ward, R. D. and Mills, C. A. (1989). A genetic stock study of perch, Perca fluviatilis L., in Windermere. J Fish Biol, 34: 965–967.
Bolten, A. B., Bjorndal, K. A., Martins, H. R. and Dellinger, T. et al1998). Transatlantic developmental migrations of loggerhead sea turtles demonstrated by mtDNA sequence analysis. Ecol Applications, 8: 1–7.
Borer, S. O., Miller, L. M. and Kapuscinski, A. R. (1999). Microsatellites in walleye Stizostedion vitreum. Mol Ecol, 8: 336–338.
Bowen, B. W., Abreu-Grobois, F. A., Balazs, G. H. and Kamezaki, N. et al1994). Trans-Pacific migrations of the loggerhead turtle (Caretta caretta) demonstrated with mitochondrial markers. Proc Natl Acad Sci USA, 92: 3731–3734.
Brown, G. E. and Brown, J. A. (1993). Do kin always make better neighbors: the effects of territory quality. Behav Ecol Sociobiol, 33: 225–231.
Brown, G. E. and Brown, J. A. (1996). Does kin-biased territorial behavior increase kin-based foraging in juvenile salmonids? Behav Ecol, 7: 24–29.
Brunner, P. C., Douglas, M. R. and Bernatchez, L. (1998). Microsatellite and mitochondrial DNA assessment of population structure and stocking effects in Arctic charr Salvelinus alpinus (Teleostei: Salmonidae) from central Alpine lakes. Mol Ecol, 7: 209–223.
Carvalho, G. R. (1993). Evolutionary aspects of fish distribution: genetic variability and adaptation. J Fish Biol, 43 (Suppl.), 53–73, 10.1006/jfbi.1993.1208.
Chapuisat, M., Goudet, J. and Keller, L. (1997). Microsatellites reveal high population viscosity and limited dispersal in the ant Formica paralugubris. Evolution, 51: 475–482.
Chenoweth, S. F., Hughes, J. M., Keenan, C. P. and Lavery, S. (1998). Concordance between dispersal and mitochondrial gene flow: isolation by distance in a tropical teleost, Lates calcarifer (Australian barramundi). Heredity, 80: 187–197.
Dieckmann, U. and Doebeli, M. (1999). On the origin of species by sympatric speciation. Nature, 400: 354–357.
Dieterich, A. (1998). Die Parasitierung der Flußbarsche (Perca fluviatilis) mit Wurmstar und Hechtbandwurm im Bodensee. MSc Thesis, Universität Rostock.
Dingle, H. (1996). Migration. Oxford University Press, New York.
Ferguson, M. M. and Noakes, D. L. G. (1981). Social grouping and genetic variation in common shiners Notropis cornutus (Pisces Cyprinidae). Environ Biol Fish, 6: 357–360.
Fitzgerald, G. J. and Morrissette, J. (1992). Kin recognition and choice of shoal mates by threespine sticklebacks. Ethol Ecol Evol, 4: 273–283.
Fontaine, P. M. and Dodson, J. J. (1999). An analysis of the distribution of juvenile Atlantic salmon (Salmo salar) in nature as a function of relatedness using microsatellites. Mol Ecol, 8: 189–198.
Goudet, J. (1995). FSTAT, version 1.2: a computer program to calculate F-statistics. J Hered, 86: 485–486.
Gyllensten, U. B. and Wilson, A. C. (1987). Mitochondrial DNA of salmonids. In: Ryman, N. and Utter, F. (eds) Population Genetics and Fisheries Management. University of Washington Press, Seattle.
Hamilton, W. D. (1964). The genetical evolution of social behaviour. I. J Theor Biol, 7: 1–16.
Hasler, A. D. and Scholz, A. T. (1983). Olfactory Imprinting and Homing in Salmon. Springer-Verlag, Berlin.
Hepper, P. G. (1986). Kin recognition: functions and mechanisms: a review. Biol Rev, 61: 63–93.
Herbinger, C. M., Doyle, R. W., Taggart, C. T. and Lochmann, S. E. et al1997). Family relationships and effective population size in a natural cohort of Atlantic cod (Gadus morhua) larvae. Can J Fish Aquat Sci, 54 (Suppl. 1) 11–18.
Imbrock, F., Appenzeller, A. and Eckmann, R. (1996). Diet and seasonal distribution of perch in Lake Constance: a hydroacoustic study and in situ observations. J Fish Biol, 49: 1–13.
Jennings, M. J., Claussen, J. E. and Philipp, D. P. (1996). Evidence for heritable preferences for spawning habitat between two walleye groups. Trans Amer Fish Soc, 125: 978–982.
Kalleberg, H. (1958). Observations in a stream tank of territoriality and competition in juvenile salmon and trout (Salmo salar & S. trutta). Rep Inst Freshwater Res, Drottninholm, 39: 55–98.
Keenleyside, G. J. A. and Yamamoto, F. T. (1962). Territorial behaviour of juvenile Atlantic salmon (Salmo salar L.). Behaviour, 19: 139–169.
Kiefer, F. (1972). Naturkunde des Bodensees, Jan Thorbecke-Verlag, Sigmaringen.
Kipling, C. and Le Cren, E. D. (1984). Mark–recapture experiments on fish in Windermere. J Fish Biol, 24: 395–414.
Kondrashov, A. S. (1998). On the origin of species by means of assortative mating. Proc R Soc B, 265: 2273–2278.
Kondrashov, A. S. and Kondrashov, F. A. (1999). Interactions among quantitative traits in the course of sympatric speciation. Nature, 400: 351–354.
Mantel, N. (1967). The detection of disease clustering as a generalized regression approach. Cancer Res, 27: 209–220.
Michalakis, Y. and Excoffier, L. (1996). A genetic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics, 142: 1061–1064.
Nepszy, S. J., Davies, D. and Einhouse, D. (1991). Walleye in Lake Erie and Lake St Clair. In: Colby, C. A., Lewis, C. A. and Eschenroder, R. L. (eds) Status of the Walleye in the Great Lakes: Case Studies Prepared for the 1989 Workshop, pp. 145–568. Great Lakes Fishery Commission Special Publication, 91–1, Ann Arbor, MI.
Pamilo, P. (1984). Genotypic correlation and regression in social groups: multiple alleles, multiple loci and subdivided populations. Genetics, 107: 307–320.
Pamilo, P. (1985). Effect of inbreeding on genetic relatedness. Hereditas, 103: 195–200.
Pamilo, P. (1989). Estimating relatedness in social groups. Trends Ecol Evol, 4: 353–355.
Piertney, S. B., Maccoll, A. D. C., Lambin, X. and Moss, R. et al, 1999). Spatial distribution of genetic relatedness in a moorland population of red grouse (Lagopus lagopus scoticus). Biol J Linn Soc, 68: 317–331.
Pitcher, T. J. (1986). The Behaviour of Teleost Fishes. Croom Hell, London.
Pouyaud, L., Desmarais, E., Chenuil, A. and Agnese, J. F. et al1999). Kin cohesiveness and possible inbreeding in the mouthbrooding tilapia Sarotherodon melanotheron (Pisces: Cichlidae). Mol Ecol, 8: 803–812.
Queller, D. C. and Goodnight, K. F. (1989). Estimating relatedness using genetic markers. Evolution, 43: 258–275.
Quinn, T. P. and Busack, C. A. (1985). Chemosensory recognition of siblings in juvenile coho salmon (Oncorhynchus kisutch). Anim Behav, 33: 51–56.
Quinn, T. P. and Hara, T. J. (1986). Sibling recognition and olfactory sensitivity in juvenile coho salmon (Oncorhynchus kisutch). Can J Zool, 64: 921–925.
Raymond, M. and Rousset, F. (1995). GENEPOP: a population genetics software for exact test and ecumenicism. J Hered, 86: 248–249.
Reynolds, J., Weir, B. S. and Cockerham, C. C. (1983). Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics, 195: 767–779.
Rice, W. R. (1989). Analyzing tables of statistical tests. Evolution, 43: 223–225.
Sambrook, J., Fritsch, E. F. and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York.
Sas Institute, I. S. (1986). Supplemental library user’s guide.
Sas Institute, I. S. (1995). JMP statistics and graphics guide (version 3.1). Cary, NC.
Schneider, S., Kuffer, J. M., Roessle, D. and Excoffier, L. (1997). ARLEQUIN, version 1.1: A software for population genetic data analysis. Genetic and Biometry Laboratory, University of Geneva, Switzerland.
Sinclair, M. (1988). Marine populations: An Essay on Population Regulation and Speciation. Washington University Press, Seattle.
Slatkin, M. (1993). Isolation by distance in equilibrium and nonequilibrium populations. Evolution, 47: 264–279.
Stabell, O. B. (1987). Intraspecific pheromone discrimination and substrate marking by Atlantic salmon parr. J Chem Ecol, 13: 1625–1643.
Stepien, C. A. and Faber, J. E. (1998). Population genetic structure, phylogeography and spawning philopatry in walleye (Stizostedion vitreum) from mitochondrial DNA control region sequences. Mol Ecol, 7: 1757–1769.
Tessier, N., Bernatchez, L. and Wright, J. M. (1997). Population structure and impact of supportive breeding inferred from mitochondrial and microsatellite DNA analyses in land-locked Atlantic salmon Salmo salar L. Mol Ecol, 6: 735–750.
Treasure, J. W. (1981). Some aspects of the reproductive biology of perch Perca fluviatilis L. Fecundity, maturation and spawning behaviour. J Fish Biol, 18: 729–740.
van Oppen, M. J. H., Turner, G. F., Rico, C. and Robinson, R. L. et al1998). Assortative mating among rock-dwelling cichlid fishes supports high estimates of species richness from Lake Malawi. Mol Ecol, 7: 991–1001.
Wang, N. and Eckmann, R. (1994). Distribution of perch (Perca fluviatilis L.) during their first year of life in Lake Constance. Hydrobiologia, 277: 135–143.
Weir, B. S. (1990). Genetic Data Analysis. Sinauer, Sunderland, MA.
Willemsen, J. (1977). Population dynamics of percids in Lake IJssel and some smaller lakes in the Netherlands. J Fish Res Board Can, 34: 1710–1719.
Wright, S. (1969). The foundations of population genetics. In: Brink, R. A. (ed.) Heritage from Mendel. University of Wisconsin Press, Madison, WI.
Acknowledgements
We thank Willi Nagl and Jerome Goudet for statistical advice and Jelle Atema, David Reznick, Tomas Hrbek, Tony Wilson, John Brookfield and two unknown referees for helpful comments on the manuscript. We are indebted to private fishermen and the Limnological Institute of the University of Konstanz for logistical support. This research was supported by the Margarete von Wrangell Habilitation Programm, the Deutsche Forschungsgemeinschaft DFG, the Verband der Chemischen Industrie and the University of Konstanz.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gerlach, G., Schardt, U., Eckmann, R. et al. Kin-structured subpopulations in Eurasian perch (Perca fluviatilis L.). Heredity 86, 213–221 (2001). https://doi.org/10.1046/j.1365-2540.2001.00825.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.2001.00825.x
Keywords
This article is cited by
-
The influence of feeding ecology and location on total mercury concentrations in Eurasian perch (Perca fluviatilis)
Environmental Sciences Europe (2021)
-
Twelve new microsatellite loci of Eurasian perch Perca fluviatilis Linnaeus, 1758
Biologia Futura (2021)
-
Phenotypic assortment by body shape in wild-caught fish shoals
The Science of Nature (2018)
-
Two decades of genetic consistency in a reproductive population in the face of exploitation: patterns of adult and larval walleye (Sander vitreus) from Lake Erie’s Maumee River
Conservation Genetics (2016)
-
Diet, stable isotopes and morphology of Eurasian perch (Perca fluviatilis) in littoral and pelagic habitats in the northern Baltic Proper
Environmental Biology of Fishes (2014)
