
Abstract
Diabetic retinopathy is a leading cause of acquired visual loss. Current treatment modalities are not effective in all cases and may have side effects. Investigation of the biochemical basis of diabetic retinopathy suggests that future treatments may reverse or halt the progression of diabetic retinopathy, or actually prevent the development of diabetic retinopathy. Pharmacological manipulation of protein kinase C and various growth factors may form the basis of future treaments for diabetic retinopathy.
Similar content being viewed by others

Introduction
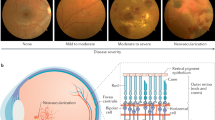
Diabetic retinopathy (DR) remains a significant cause of acquired visual loss in adults worldwide and it is the number one cause of new blindness in the working aged population in western countries, thus leading to substantial medical, social, and financial burdens.1 In the United States, diabetes mellitus affects over 16 million people, although only half of this number are aware that they have the disease. Similarly, at least 2% of the United Kingdom population is known to have the disease and in both countries DR is the leading cause of new cases of legal blindness in adults between 20 and 70 years of age.2 Some level of DR is present in 2.5% of the population of the United States (Vision Problems in the US, available from National Eye Institute and Research to Prevent Blindness, 2002) and in nearly all diabetic patients with duration of diabetes of 20 years or more. Approximately 25% of patients have the more severe sight-threatening levels of retinopathy.3,4
Severe visual loss in patients with diabetes primarily arises from intraocular angiogenesis resulting in proliferative diabetic retinopathy (PDR), while the main cause of moderate visual loss is leakage of retinal vessels resulting in diabetic macular oedema (DME). Since the great majority of patients have type II diabetes and since DME is more common in type 2 disease, more people have visual loss from DME than from PDR.5 The risk of severe visual loss, defined as visual acuity of 5/200 or worse at two consecutive visits 4 months apart, from PDR is about 60% after 6 years if not treated with laser panretinal photocoagulation (PRP).6 The risk of moderate visual loss, which is defined as doubling of the visual angle at two consecutive visits 4 months apart, from clinically significant macular edema (ME) is about 33% after 3 years.7
Current treatment modalities can greatly reduce this visual loss; however, these modalities are not successful in every case and may be associated with significant side effects and complications. Owing to the limitations, potential side effects, and complications of these treatments, significant efforts continue to be directed towards the development of novel, more effective, and nondestructive therapeutic modalities. Investigations into such potential therapies have taken a variety of approaches and include optimization of systemic conditions including blood glucose levels and hypertension, blockade of angiogenic growth factors, blockade of intracellular signalling and pathways utilized by these angiogenic agents, evaluation of naturally occurring endogenous angiogenic inhibitors, evaluation of the role of the extracellular matrix and vitreo-retinal interface, and antioxidants.
Current treatment for DR
Two large multicentre, prospective, case–control studies, the Diabetic Retinopathy Study (DRS)6 and the Early Treatment of Diabetic Retinopathy Study (ETDRS),7 provided the evidence that laser photocoagulation reduces the risk of visual loss in DR. Specifically, the DRS demonstrated that PRP reduced the risk of severe visual loss by 50% in eyes with high-risk proliferative diabetic retinopathy (HRC PDR).6 The ETDRS demonstrated that early detection of the development of such high-risk characteristics of retinal neovascularization and timely PRP reduced the risk of severe visual loss to less than 2%.7 The ETDRS also defined clinically significant macular edema (CSME) as being ME involving or threatening the centre of the macula and demonstrated that focal or grid laser photocoagulation could reduce the risk of moderate visual loss due to CSME by 50%.7
The comprehensive nature of this evidence has led to the widespread incorporation of these treatment approaches into standard clinical practice. Laser treatment of eyes at or approaching HRC-DR and/or CSME is the current standard of care. However, laser photocoagulation has potential side effects, including reduction in visual field and nyctalopia arising from the inherent destruction of retinal tissue associated with PRP. Potential complications include damage to other intraocular structures or misplaced burns. Furthermore, the chorioretinal scars associated with laser photocoagulation can enlarge over time, resulting in foveal encroachment or extensive photoreceptor loss, pigment clumping, and vessel attenuation, mimicking that observed in retinitis pigmentosa.
Optimization of glycaemic control and systemic hypertension
Two important multicentre clinical trials, the Diabetes Control and Complications Trial (DCCT) and the United Kingdom Prospective Diabetes Study (UKPDS), conclusively demonstrated that intensive glycaemic control slows the onset and progression of DR and other vascular complications in both type 1 and type 2 diabetes.8,9,10 A total of 1441 patients with type 1 DM was studied longitudinally in the DCCT for a mean duration of 6.5 years. The mean HbA1c in the intensive therapy group was significantly less than that in the conventional therapy group (7.2 and 9.1%, respectively).8 Intensive therapy resulted in a 76% reduction in the onset of DR, a 63% reduction in the progression of DR, a 47% reduction in the development of severe nonproliferative or proliferative DR, a 26% reduction in the development of macular edema, and a 51% reduction in the overall need for laser.8,11 The disadvantages of intensive insulin therapy included increased episodes of hypoglycaemia and a two times greater occurrence of early initial worsening in retinopathy, which was reversed after 18 months.12 This reduction in the risk of progression of retinopathy resulting from intensive insulin therapy persisted for at least 4 years after the end of the DCCT trial, despite the fact that 4 years after the end of DCCT the levels of glycaemic control converged for both groups.9
The multicentre UKPDS study involved 3867 patients with newly diagnosed type 2 DM, who were randomly assigned to intensive therapy (using either sulphonylurea or insulin) or conventional control.10 The mean HbA1c after 10 years was significantly lower in the intensive therapy group than in the conventional therapy group (7.0 and 7.9%, respectively). Intensive glycaemic control significantly reduced the need for retinal photocoagulation by 29% and resulted in a 17% reduction in the progression of retinopathy, both of which were significant when compared with the conventional treatment group.13 Similar risk reductions were observed using either sulphonylureas or insulin therapy.
It has been demonstrated that patients with diabetes have a higher prevalence and incidence of systemic hypertension than people without diabetes.14 Elevated blood pressure (BP) is associated with more severe levels of retinopathy initially, an increased risk of developing any retinopathy, an increased risk of developing more severe levels of retinopathy, and an increased risk of more rapid progression of retinopathy when compared to diabetic patients without hypertension.15,16,17 A recent review of systemic factors influencing DR highlighted that the two main types of epidemiological studies of the relation between DR and hypertension were cross-sectional studies and prospective longitudinal studies. Overall, about two-thirds of cross-sectional reports of both type 1 and type 2 diabetic patients over the last 20 years demonstrated an association between DR and hypertension, but this association was weak in elderly type 2 patients. Interestingly, only one of the more recent prospective studies did not demonstrate an association between elevated BP and DR.18
The UKPDS also demonstrated that elevated BP was an independent risk factor for onset and progression of DR.19 Of 1148 patients enrolled into this part of the study, 758 patients were assigned to an intensive BP control group and 390 patients were assigned to a less tight control group. A significant BP reduction was achieved by the intensive group over a median of 8.4 years resulting in a 34% reduction in DR progression and 47% reduction in moderate visual acuity loss, both of which were significantly lower than the reduction seen in patients with moderate BP control.19 These risk reductions were independent of glycaemic control and were similar, regardless of whether the hypertension was controlled with angiotensin-converting enzyme (ACE) inhibitors or beta blockers.
Recent laboratory studies have suggested a number of mechanisms by which hypertension may increase the risk of developing DR.20 These mechanisms include hypertension-induced vascular stretching resulting in increased expression of vascular endothelial growth factor (VEGF) and its receptors,21 and enhancement of retinal endothelial damage.22 While epidemiological and laboratory studies into the effects and mechanisms of elevated BP in DR are ongoing, there is sufficient evidence available that all diabetic patients should have their BP maintained as low as safely possible.
Blockade of angiogenic factors
Recently, there has been an explosion in knowledge regarding the biochemical processes underlying DR. Particular attention has been focused on the role of growth factors in the pathogenesis of DR. Ironically, Michaelson23 first proposed the concept of growth factors 50 years ago, and this concept has been elaborated further by subsequent authors.24 The theory predicts ideal characteristics that a molecule should possess if it truly caused proliferative retinopathy. These characteristics included production of the molecule by the retina, induction by hypoxia, stimulation of neovascularization, and secretion into and diffusion throughout the eye. Endothelial cells would be expected to have high-affinity receptors for such a molecule, intraocular levels should be elevated during proliferation, reduced after treatment or during quiescence, and blockage of its effects should suppress hypoxia-induced neovascularization. Demonstration of these criteria in a candidate growth factor provides strong evidence that it could serve as a major mediator of proliferative retinopathy.
Numerous candidate molecules have been investigated as potential mediators of diabetic retinal vascular complications. Those that have received considerable evaluation include basic fibroblastic growth factor (bFGF), growth hormone (GH),25,26 insulin-like growth factor -1 (IGF-1),27,28 connective tissue growth factor (CTGF),29 prostacyclin-stimulating factor (PSF),30 hepatocyte growth factor (HGF),31,32,33 and vascular endothelial growth factor (VEGF).34,35 bFGF, GH, IGF-1, and CTGF are growth factors that do not fulfil all of the Michaelson criteria, but probably have either synergistic or permissive roles in PDR. The expression of these permissive or synergistic growth factors may be regulated by a complex interaction of different growth factors, which is still not fully understood. For example, bFGF acts synergistically with VEGF,36 but unlike VEGF is not primarily responsible for ischaemia-induced retinal neovascularization.37 Another example is CTGF, which may have a role in the pathogenesis of retinal fibrosis, matrix deposition, and angiogenesis. Retinal vascular endothelial cells express this molecule and VEGF appears to increase CTGF gene expression in endothelial cells via VEGF receptors and the activation of PI-3 kinase but not protein kinase C (PKC) or RAS pathways.38 Another factor, PSF, stimulates endothelial cell synthesis of prostacyclin PG12, a vasoactive prostaglandin, which may be responsible for the biphasic changes in retinal blood flow seen in diabetes.39
HGF has attributes consistent with a mediator of proliferative retinopathy, but its role is still under investigation. HGF is a pleiotropic molecule and its receptor, c-Met, like that of VEGF's KDR receptor, is a transmembrane tyrosine kinase that is autophosphorylated in response to HGF binding.40,41 HGF acts as a mitogen in many cells, including retinal endothelial cells, which are sensitive to the effects of HGF at physiologically relevant concentrations.42 There is evidence that the HGF/cMet signal transduction cascade involves PI3 kinase, PKC, and MAP kinase pathways, and HGF in vivo can increase retinal permeability.
VEGF is the most extensively studied molecule that fulfils all of the Michaelson criteria. For this reason, it is a principal focus of novel treatments for PDR, other proliferative retinopathies, and systemic diseases characterized by abnormal angiogenesis. VEGF is a highly conserved glycoprotein with four isoforms, the two smaller of which are freely diffusible within the eye.43 VEGF is produced by many intraocular cells including pigment epithelial cells, pericytes, and endothelial cells.44,45 Autophosphorylating tyrosine kinase VEGF receptors (KDR receptor) have been identified on retinal microvascular endothelial cells at high densities.46 VEGF is primarily an endothelial mitogen,43,47,48 and animal models 49,50,51,52,53 and clinical studies34,54,55,56 have demonstrated hypoxia-induced retinal VEGF concentration elevations leading to retinal neovascularization. Furthermore, intraocular levels of VEGF decrease after adequate treatment of retinal neovascularization with laser photocoagulation. Increased vascular permeability is another hallmark of intraocular neovascularization, and direct evidence now exists that physiologic concentrations of VEGF increase retinal vascular permeability as measured by vitreous fluorophotometry and that this increased permeability is suppressed by VEGF inhibitors.57
Biochemically, hypoxia-induced increases in adenosine can bind to the adenosine A2 receptor and potentially mediate hypoxia-induced VEGF expression, while synergistically inducing endothelial migration (through A2 receptors) and capillary tube formation (through A1 and A2 receptors).58,59 Reactive oxygen intermediates also stimulate VEGF mRNA and protein production, an effect that is dose-dependent and blocked by antioxidants.60 Experimental inhibition of VEGF production or binding to its receptor results has an antiproliferative effect demonstrated using a number of different agents in several different animal models. In a primate model, iris neovascularization from laser-induced retinal vein occlusion was reduced by VEGF-neutralizing monoclonal antibody.61 In a mouse neovascularization model, both chimeric proteins (which sequestered VEGF)62 and antisense oligonucleotides (which suppress VEGF transcription) reduced retinal new vessel formation by 40–100%.63 It has also been demonstrated in a murine model of retinal neovascularization that inhibition of VEGF receptor kinase activity completely blocks retinal neovascularization.64
Blockade of signal transduction pathways
The signal transduction mechanism initiated by VEGF binding to its receptors on retinal endothelial cells is characterized by increased tyrosine phosphorylation of phospholipase γ (PLC γ) and PI-3 kinase.65 PLC γ activation leads to increases in diacylglycerol and subsequent activation of classical forms of PKC, especially the βII and δ isoforms.66
Experimental work has demonstrated that selective inhibition of the PKC β isoform prevents VEGF-mediated cell growth in vitro,57 while inhibition of the PKC α isoform stimulates growth.57 Furthermore, inhibition of PKC β in vivo reduces ischaemic retinal neovascularization in an animal model.67 Both intravitreal and oral forms of a PKC β inhibitor have been shown to block VEGF-induced increases in retinal vascular permeability in animals and to normalize changes in retinal blood flow characteristically induced by DR in both animals and humans.68 Recent studies also demonstrate that PKC inhibition can normalize diabetes-induced retinal vascular permeability in animals, even after diabetes has been established for as long as 1 month prior to initiating therapy (Aiello, unpublished data).
Recent data derived from transgenic mouse models provide substantial support for the hypothesis that PKC β is involved in mediating retinal neovascularization and that inhibition of this molecule may have therapeutic benefit. Transgenic animals were derived where PKC β2 is overexpressed in the vasculature using a pre-pro-endothelin promoter. When these animals experience retinal ischaemia, the retinal neovascular response is greatly accentuated, suggesting that PKC β expression plays a significant role in this response. More important information from a therapeutic standpoint is derived from animals where the PKC β gene has been removed, resulting in animals with no PKC β (PKC β knockout). When PKC β knockout mice are exposed to similar retinal ischaemia, retinal neovascularization is reduced.69
These data and the safety and pharmocologic results of PKC β inhibitors to date have supported the undertaking of two Phase II/III studies and a large Phase III study. These two Phase II/III studies have just recently been completed and data analysis should begin soon. These studies evaluate whether an orally administered PKC β selective inhibitor is effective in preventing the progression of nonproliferative DR to PDR or in preventing progression (or inducing regression) of DME. To date, the safety profile of the medication has been excellent and no clinically significant side effects associated with the compound have been appreciated.
Endogenous inhibitors of angiogenesis
Neovascularization probably results from an alteration of the normal balance between natural promoters and natural inhibitors of angiogenesis in favour of proangiogenic molecules. A number of molecules have been identified as endogenous inhibitors of angiogenesis including endostatin, angiostatin, thrombospondin-1, and pigment epithelium-derived factor (PEDF). Of these natural antiangiogenic agents, PEDF has become a major focus of attention.70
PEDF is a 50-kDa molecule, originally purified from human retinal pigment epithelial cells, which contains protein sequences homozogous to the serine protease inhibitor (SERPIN) family of molecules.70 PEDF, however, does not inhibit serine proteases. Recent experimental work has shown that PEDF can inhibit VEGF, bFGF, and other angiogenic factors in vitro, an effect reversed by antibody neutralization of PEDF.70 The amount of PEDF produced by retinal cells was positively correlated with oxygen concentrations in animal models, while hypoxia-induced retinal neovascularization resulted in a 10-fold increase in the VEGF/PEDF ratio in a rat model.71 PEDF has also been shown to inhibit hypoxia-induced angiogenesis in a mouse model of retinal neovascularization.64,72 As understanding of the mechanism of action of PEDF increases, future novel treatments of DR might utilize synthetic versions of this molecule or stimulate similar intracellular pathways to prevent the onset of intraocular angiogenesis.
Alterations of the extracellular matrix and vitreo-retinal interface
Remodelling of the extracellular matrix, which enables endothelial cells to migrate towards an angiogenic stimulus, is a component of proliferative retinopathies. Two main groups of molecules are involved in this migration of endothelial cells. The first of these are the matrix metalloproteinases (MMPs), a family of zinc-binding, calcium-dependent enzymes that selectively degrade the extracellular matrix. MMPs are inhibited naturally by a separate group of multifunctional proteins known as tissue inhibitors of metalloproteinases (TIMP). A balance between MMPs and TIMPs probably preserves tissue integrity in normal eyes, as evidenced by expression of MMP-1 (interstitial collagenase) and TIMP-2 in nondiseased retina.73
Increased levels of MMP-9 (gelatinase B) have been demonstrated in the vitreous of eyes with PDR, while fibrovascular membranes from eyes with PDR stain for MMP-1, MMP-2 (gelatinase A), MMP-3 (stromelysin-1), MMP-9, TIMP-1, TIMP-2, and TIMP-3. Release of both MMPs and TIMPs may be under the control of cytokines produced by a number of ocular cells, including endothelial cells, although the exact mechanisms are not fully understood.74,75 Future novel therapies for proliferative retinopathies, including DR, may be directed at manipulating the intraocular balance between MMPs and TIMPs.76
Integrins are another group of molecules that mediate migration of vascular endothelial cells through the extracellular matrix. These are cell-surface adhesion molecules that interact with numerous components of the extracellular matrix.77 Two integrins in particular, 2vβ3 and 2vβ5, have been studied with regard to intraocular neovascularization and appear to mediate two separate cytokine-dependent angiogenesis pathways.77 Integrin 2vβ3 mediates one pathway induced by bFGF or tissue necrosis factor α (TNFα),78 and promotes growth by suppression of apoptosis and upregulation of angiogenesis promoter p53.79 Integrin 2vβ5 mediates a second pathway induced by VEGF or transforming growth factor α (TGFα), which is PKC dependent.78 Both integrins 2vβ3 and 2vβ5 are detectable in fibrovascular membranes of eyes with PDR.77 Systemically administered cyclic peptides, which block the integrins, prevent retinal angiogenesis in a mouse model,77 and can inhibit hypoxia-induced retinal neovascularization.80 Inhibition of integrins 2vβ3 and 2vβ5 by such peptides or molecules that mimic their action could provide another target for future novel therapies for DR and other proliferative retinopathies.
Some of the complications of PDR are a result of the interaction between the vitreous and retinal surface new vessels. Intraocular injections of enzymes such as plasmin and chondroitinase to cleave structural components of the vitreous body or inner retinal surface have been demonstrated to create posterior vitreous detachment (PVD) in animals81,82,83 and human eyes.84 The ability to create a PVD when required would be advantageous in diabetic patients as attached vitreous provides scaffolding which can support retinal neovascularization. Prophylactic removal of such support would also prevent vitreous traction on retinal new vessels, potentially reducing the risk of vitreous haemorrhage and tractional retinal detachment. A similar approach to speed resolution of vitreous haemorrhage is already approved in Mexico and pending evaluation in the US.
Antioxidants
Another possible novel preventative oral therapy for DR is vitamin E. Studies of vitamin E in diabetic animals have shown reduced retinal DAG levels, normalized PKC-β activation, normalized retinal blood flow, and restored nitric oxide-mediated endothelium-dependent relaxation.85,86 A recent clinical study demonstrated that short-term, high-dose oral vitamin E therapy normalized retinal and renal haemodynamics in diabetic patients with disease of less than 10 years duration despite no change in glycaemic control.87 Whether these changes will eventually result in suppression of DR awaits a prospective, randomized clinical trial. Since overdosage with vitamin E can potentially result in toxicity, high doses of vitamin E should not be advocated until results of appropriate clinical trials are known.
Changing the treatment paradigm for novel therapies
At present, PRP is indicated for eyes with HRC PDR and should be considered for eyes approaching this stage. Macular focal laser photocoagulation is indicated for patients with CSME. This treatment paradigm balances the threat of visual loss against the potential damaging effects of laser therapy. Treatment is applied at stages of DR where the underlying biochemical processes are already advanced. In some instances, these changes may be irreversible, as evidenced by cases that fail to respond to laser therapy despite timely intervention.
Novel therapies that manipulate the balance between proangiogenic molecules like VEGF and antiangiogenic molecules like PEDF have significant and partially opposing implications for the microvascular and macrovascular complications of diabetes.88 In microvascular complications such as DR and diabetic nephropathy, production of VEGF leads to damaging neovascularization and increased retinal permeability. In macrovascular complications such as peripheral vascular disease and coronary artery disease, the same hypoxia-induced VEGF expression beneficially promotes collateral circulation leading to reperfusion of ischaemic tissue. Prospective novel treatments face the paradox of using antiangiogenic agents to treat DR that may decrease healing in peripheral limb ischaemia or proangiogenic agents to treat peripheral vascular disease that may detrimentally alter the ocular state.89 Interestingly, therapeutic local injections of VEGF cDNA into muscle increase perfusion of ischaemic areas with activity remaining primarily local, presumably because of the rapid degradation of VEGF in the bloodstream. Furthermore, as mentioned above, trials of systemic inhibitors of VEGF have not been associated with significant systemic side effects, suggesting that these separate treatment paradigms may not necessarily be mutually exclusive.
Laboratory and clinical research is now focused on preventing the development of retinal neovascularization and pathologic retinal vascular leakage, reversing or retarding the progression of retinal neovascularization and pathologic retinal vascular leakage, correcting the underlying disease mechanisms, and restoring damaged tissue. Several clinical trials to accomplish these goals, using either an intravitreal administration approach or nonocular administration approach, have been completed, are underway, or are planned.
For DME, intravitreal steroid sustained release implant (fluorcinolone acetonide), intravitreal steroid biodegradable sustained release implant (dexamethasone), and intravitreal aptamer VEGF inhibitor (macugen) are currently undergoing clinical trial for ocular complications of diabetes. Additionally, studies using intravitreal steroid injections (triamcinolene acetonide) and intravitreal humanized anti-VEGF antibody fragment (rhuFab V2) to treat DMF are in the planning stage.
For nonocular administration approaches, Phase 1/2 clinical trials of an oral, once daily PKC β inhibitor (LY333531) to treat nonproliferative and proliferative DR and DME have been recently completed. A Phase III trial is ongoing. Trials with an intramuscular injection of a growth hormone antagonist (Sandostatin) to treat PDR and an oral COX2 inhibitor (Celebrex) to treat DME are also ongoing.
With the arrival of novel therapies for the treatment of DR, monitoring the level of DR will be of paramount importance to determine the need for treatment, when to discontinue treatment, and whether to reinstitute treatment. Novel therapies with less-damaging side effects than laser therapy might be initiated at earlier stages of DR when the underlying processes are beginning and advanced tissue damage is minimal. Since the stages of DR and DME most amenable to treatment are usually asymptomatic, patients will need to be identified by regular comprehensive examinations. Novel therapies might also be used as adjuvants to laser therapy, either before laser, simultaneously with laser, or in cases where laser fails. Similarly, novel therapies might be used before or after vitrectomy or might be implanted intraocularly during vitrectomy.
Conclusion
Laser photocoagulation is the current standard of care for the treatment of sight-threatening DR. Use of this therapy is based on evidence from prospective, multicentre, randomized studies performed and analysed over the past 30 years. Laser photocoagulation is directed towards inducing the regressing of existing neovascularization or ME rather than treating the underlying biochemical processes of DR. In addition, laser photocoagulation has a number of significant side effects and potential complications. Increased understanding of the biochemical pathways underlying DR has resulted in the identification of numerous potential novel therapies. Currently, the most promising among these are inhibition of intraocular growth factors (such as VEGF), inhibition of intracellular signalling cascades (such as PKC β inhibitors), use of endogenous inhibitors (such as PEDF), and prophylactic treatment with antioxidants. In the near future, it is possible that novel therapies may result in alterations of the current treatment paradigm for DR. Treatments directed against the biochemical pathways underlying DR may best be initiated before sight-threatening levels of retinopathy are reached so as to provide optimal benefit. As early stages of DR are usually asymptomatic, patients will need to undergo regular examinations to identify the need for treatment. Overall, with the diverse array of novel therapeutic approaches currently under investigation, it is likely that the next decade will be marked by exciting new treatment options providing new benefits to our patients with diabetes and diabetic eye disease.
References
National Diabetes Data Group. Diabetes in America, 2nd ed. NIH Publ. No. 95-1468. US Govt. Printing Office: Washington, 1995.
Chief Medical Officer. The Annual Report of the Chief Medical Officer of the Department of Health for the Year 1997. Her majesty's Stationary Office: London, 2002.
Klein R, Klein BE, Moss SE . Visual impairment in diabetes. Ophthalmology 1984; 91: 1–9.
Klein R, Klein BE, Moss SE, Cruickshanks KJ . The Wisconsin epidemiologic study of diabetic retinopathy. XV. The long-term incidence of macular edema. Ophthalmology 1995; 102: 7–16.
Klein R, Klein BE, Moss SE, Davis MD, DeMets DL . The Wisconsin epidemiologic study of diabetic retinopathy. IV. Diabetic macular edema. Ophthalmology 1984; 91: 1464–1474.
The Diabetic Retinopathy Study Research Group. Photocoagulation treatment of proliferative diabetic retinopathy. Clinical application of diabetic retinopathy study (DRS) findings, DRS Report Number 8. Opthalmology 1981; 88: 583–600.
The Early Treatment Diabetic Retinopathy Study Research Group. Early photocoagulation for diabetic retinopathy. ETDRS report number 9. Ophthalmology 1991; 98: 766–785.
The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus (see comments). N Engl J Med 1993; 329: 977–986.
Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group (published erratum appears in N Engl J Med 2000; 342(18): 1376). N Engl J Med 2000; 342: 381–389.
Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group (published erratum appears in Lancet 1999 Aug 14; 354(9178): 602) (see comments). Lancet 1998; 352: 837–853.
The Diabetes Control and Complications Trial Research Group. The effect of intensive diabetes treatment on the progression of diabetic retinopathy in insulin-dependent diabetes mellitus: the diabetes control and complications trial. Arch Ophthalmol 1995; 113: 36–51.
Early worsening of diabetic retinopathy in the Diabetes Control and Complications Trial (published erratum appears in Arch Ophthalmol 1998; 116(11): 1469). Arch Ophthalmol 1998; 116: 874–886.
Intensive blood–glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group (see comments). Lancet 1998; 352: 837–853.
Klein R, Klein BE, Lee KE, Cruickshanks KJ, Moss SE . The incidence of hypertension in insulin-dependent diabetes. Arch Intern Med 1996; 156: 622–627.
Diabetes Drafting Group. Prevalence of small vessel and large vessel disease in diabetic patients from 14 centres: The World Health Organisation multinational study of vascular disease in diabetes. Diabetologia 1985; 28: 615–640.
Klein R, Klein BE, Moss SE, Cruickshanks KJ . The Wisconsin epidemiologic study of diabetic retinopathy: XVII. The 14-year incidence and progression of diabetic retinopathy and associated risk factors in type 1 diabetes (see comments). Ophthalmology 1998; 105: 1801–1815.
Marshall G, Garg SK, Jackson WE, Holmes DL, Chase HP . Factors influencing the onset and progression of diabetic retinopathy in subjects with insulin-dependent diabetes mellitus. Ophthalmology 1993; 100: 1133–1139.
Aiello LP, Cahill M, Wong JS . Systemic considerations in the management of diabetic retinopathy. Am J Ophthalmol 2001; 760–766.
Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38 (in process Citation). BMJ 1998; 317: 703–713.
Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. UK Prospective Diabetes Study Group (see comments). BMJ 1998; 317: 713–720.
Suzuma I, Hata Y, Rook SL, Suzuma K, Clermont A, Feener EP et al. Cyclic stretch and hypertension induce retinal VEGF-R2 (KDR) expression: potential mechanisms for exacerbation of diabetic retinopathy by hypertension. Diabetes 2001; 50: 444–454.
Hsueh WA, Anderson PW . Hypertension, the endothelial cell, and the vascular complications of diabetes mellitus (clinical conference). Hypertension 1992; 20: 253–263.
Michaelson IC . The mode of development of the vascular system of the retina, with some observations on its significance for certain retinal diseases. Trans Ophthalmol Soc UK 1948; 68: 137–180.
Ashton N . Retinal neovascularization in health and disease. Am J Ophthalmol 1957; 44: 7–24.
Sharp P, Fallen T, Brazier O, Sandler L, Joplin G, Kohner E . Long-term follow-up of patients who underwent Yttrium-90 pituitary implantation for treatment of proliferative diabetic retinopathy. Diabetologia 1987; 30: 199–207.
Smith LE, Kopchick JJ, Chen W, Knapp J, Kinose F, Daley D et al. Essential role of growth hormone in ischemia-induced retinal neovascularization. Science 1997; 276: 1706–1709.
Grant MB, Guay C, Marsh R . Insulin-like growth factor I stimulates proliferation, migration, and plasminogen activator release by human retinal pigment epithelial cells. Curr Eye Res 1990; 9: 323–335.
Grant MB, Mames RN, Fitzgerald C, Ellis EA, Caballero S, Chegini N et al. Insulin-like growth factor I as an angiogenic agent. In vivo and in vitro studies. Ann N Y Acad Sci 1993; 692: 230–422.
Wunderlich K, Senn BC, Todesco L, Flammer J, Meyer P . Regulation of connective tissue growth factor gene expression in retinal vascular endothelial cells by angiogenic growth factors. Graefes Arch Clin Exp Ophthalmol 2000; 238: 910–915.
Hata Y, Clermont A, Yamauchi T, Pierce EA, Suzuma I, Kagokawa H et al. Retinal expression, regulation, and functional bioactivity of prostacyclin-stimulating factor. J Clin Invest 2000; 106: 541–550.
Nishimura M, Nakano K, Ushiyama M, Nanbu A, Ohtsuka K, Takahashi H et al. Increased serum concentrations of human hepatocyte growth factor in proliferative diabetic retinopathy. J Clin Endocrinol Metab 1998; 83: 195–198.
Nishimura M, Ikeda T, Ushiyama M, Kinoshita S, Yoshimura M . Changes in vitreous concentrations of human hepatocyte growth factor (hHGF) in proliferative diabetic retinopathy: implications for intraocular hHGF production. Clin Sci (Colch) 2000; 98: 9–14.
Cai W, Rook SL, Jiang ZY, Takahara N, Aiello LP . Mechanisms of hepatocyte growth factor-induced retinal endothelial cell migration and growth. Invest Ophthalmol Vis Sci 2000; 41: 1885–1893.
Aiello LP, A very RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders (see comments). N Engl J Med 1994; 331: 1480–1487.
Aiello LP . Clinical implications of vascular growth factors in proliferative retinopathies. Curr Opin Ophthalmol 1997; 8: 19–31.
Hata Y, Rook SL, Aiello LP . Basic fibroblast growth factor induces expression of VEGF receptor KDR through a protein kinase C and p44/p42 mitogen-activated protein kinase-dependent pathway. Diabetes 1999; 48: 1145–1155.
Ozaki H, Okamoto N, Ortega S, Chang M, Ozaki K, Sadda S et al. Basic fibroblast growth factor is neither necessary nor sufficient for the development of retinal neovascularization (see comments). Am J Pathol 1998; 153: 757–765.
Suzuma K, Naruse K, Suzuma I, Takahara N, Ueki K, Aiello LP et al. Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, flt1, and phosphatidylinositol 3-kinase-akt-dependent pathways in retinal vascular cells. J Biol Chem 2000; 275: 40725–40731.
Hata Y, Clermont A, Yamauchi T, Pierce EA, Suzuma I, Kagokawa H et al. Retinal expression, regulation, and functional bioactivity of prostacyclin-stimulating factor. J Clin Invest 2000; 106: 541–550.
Rubin JS, Bottaro DP, Aaronson SA . Hepatocyte growth factor scatter factor and its receptor, the c-met proto-oncogene product. Biochim Biophys Acta 1993; 1155: 357–371.
Matsumoto K, Nakamura T . Emerging multipotent aspects of hepatocyte growth factor. J Biochem (Tokyo) 1996; 119: 591–600.
Cai W, Rook SL, Jiang ZY, Takahara N, Aiello LP . Mechanisms of hepatocyte growth factor-induced retinal endothelial cell migration and growth. Invest Ophthalmol Vis Sci 2000; 41: 1885–1893.
Ferrara N, Houck KA, Jakeman LB, Winer J, Leung DW . The vascular endothelial growth factor family of polypeptides. J Cell Biochem 1991; 47: 211–218.
Adamis AP, Shima DT, Yeo KT, Yeo TK, Brown LF, Berse B et al. Synthesis and secretion of vascular permeability factor/vascular endothelial growth factor by human retinal pigment epithelial cells. Biochem Biophys Res Commun 1993; 193: 631–638.
Simorre-Pinatel V, Guerrin M, Chollet P, Penary M, Clamens S, Malecaze F et al. Vasculotropin-VEGF stimulates retinal capillary endothelial cells through an autocrine pathway. Invest Ophthalmol Vis Sci 1994; 35: 3393–3400.
Thieme H, Aiello LP, Takagi H, Ferrara N, King GL . Comparative analysis of vascular endothelial growth factor receptors on retinal and aortic vascular endothelial cells. Diabetes 1995; 44: 98–103.
Plate KH, Breier G, Weich HA, Risau W . Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992; 359: 845–848.
Guerrin M, Moukadiri H, Chollet P, Moro F, Dutt K, Malecaze F et al. Vasculotropin/vascular endothelial growth factor is an autocrine growth factor for human retinal pigment epithelial cells cultured in vitro. J Cell Physiol 1995; 164: 385–394.
Miller JW, Adamis AP, Shima DT, D’Amore PA, Moulton RS, O’Reilly MS et al. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am J Pathol 1994; 145: 574–584.
Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LE . Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci USA 1995; 92: 905–909.
Donahue ML, Phelps DL, Watkins RH, LoMonaco MB, Horowitz S . Retinal vascular endothelial growth factor (VEGF) mRNA expression is altered in relation to neovascularization in oxygen induced retinopathy. Curr Eye Res 1996; 15: 175–184.
Stone J, Chan-Ling T, Pe’er J, Itin A, Gnessin H, Keshet E . Roles of vascular endothelial growth factor and astrocyte degeneration in the genesis of retinopathy of prematurity. Invest Ophthalmol Vis Sci 1996; 37: 290–299.
Dorey CK, Aouididi S, Reynaud X, Dvorak HF, Brown LF . Correlation of vascular permeability factor/vascular endothelial growth factor with extraretinal neovascularization in the rat (see comments) (published erratum appears in Arch Ophthalmol 1997; 115(2): 192). Arch Ophthalmol 1996; 114: 1210–1217.
Adamis AP, Miller JW, Bernal MT, D’Amico DJ, Folkman J, Yeo TK et al. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferate diabetic retinopathy. Am J Ophthalmol 1994; 118: 445–450.
Pe’er J, Folberg R, Itin A, Gnessin H, Hemo I, Keshet E . Upregulated expression of vascular endothelial growth factor in proliferative diabetic retinopathy. Br J Ophthalmol 1996; 80: 241–245.
Malecaze F, Clamens S, Simorre-Pinatel V, Mathis A, Chollet P, Favard C et al. Detection of vascular endothelial growth factor messenger RNA and vascular endothelial growth factor-like activity in proliferate diabetic retinopathy. Arch Ophthalmol 1994; 112: 1476–1482.
Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes 1997; 46: 1473–1480.
Hashimoto E, Kage K, Ogita T, Nakaoka T, Matsuoka R, Kira Y . Adenosine as an endogenous mediator of hypoxia for induction of vascular endothelial growth factor mRNA in U-937 cells. Biochem Biophys Res Commun 1994; 204: 318–324.
Takagi H, King GL, Robinson GS, Ferrara N, Aiello LP . Adenosine mediates hypoxic induction of vascular endothelial growth factor in retinal pericytes and endothelial cells. Invest Ophthalmol Vis Sci 1996; 37: 2165–2176.
Kuroki M, Voest EE, Amano S, Beerepoot LV, Takashima S, Tolentino M et al. Reactive oxygen intermediates increase vascular endothelial growth factor expression in vitro and in vivo. J Clin Invest 1996; 98: 1667–1675.
Adamis AP, Shima DT, Tolentino MJ, Gragoudas ES, Ferrara N, Folkman J et al. Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a nonhuman primate. Arch Ophthalmol 1996; 114: 66–71.
Aiello LP, Pierce EA, Foley ED, Takagi H, Chen H, Riddle L et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA 1995; 92: 10457–10461.
Robinson GS, Pierce EA, Rook SL, Foley E, Webb R, Smith LE . Oligodeoxynucleotides inhibit retinal neovascularization in a murine model of proliferative retinopathy. Proc Natl Acad Sci USA 1996; 93: 4851–4856.
Ozaki H, Seo MS, Ozaki K, Yamada H, Yamada E, Okamoto N et al. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am J Pathol 2000; 156: 697–707.
Xia P, Aiello LP, Ishii H, Jiang ZY, Park DJ, Robinson GS et al. Characterization of vascular endothelial growth factor's effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J Clin Invest 1996; 98: 2018–2026.
Aiello LP, Hata Y . Molecular mechanisms of growth factor action in diabetic retinopathy. Curr Opin Endocrinol and Diabetes 1999; 6: 146–156.
Danis RP, Bingaman DP, Jirousek M, Yang Y . Inhibition of intraocular neovascularization caused by retinal ischemia in pigs by PKCbeta inhibition with LY333531. Invest Ophthalmol Vis Sci 1998; 39: 171–179.
Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor (see comments). Science 1996; 272: 728–731.
Suzuma K, Takahara N, Suzuma I, Isshiki T, Ueki K, Leitges M et al. Characterization of protein kinase C beta isoform's action on retinoblastoma protein phosphorylation, vascular endothelial growth factor-induced endothelial cell proliferation, and retinal neovascularization. Proc Natl Acad Sci USA 2002; 99: 721–726.
Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu H, Benedict W et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 1999; 285: 245–248.
Gao G, Li Y, Zhang D, Gee S, Crosson C, Ma J et al. Unbalanced expression of VEGF and PEDF in ischemia-induced retinal neovascularization. FEBS Lett 2001; 489: 270–276.
Duh E, Yang HS, Suzuma I, Miyagi M, Youngman E, Mori K et al. Pigment epithelium-derived factor suppresses ischemia-induced retinal neovascularization and VEGF-induced migration and growth. Invest Ophthalmol Vis Sci 2002; 43: 821–829.
Webster L, Chignell AH, Limb GA . Predominance of MMP-1 and MMP-2 in epiretinal and subretinal membranes of proliferative vitreoretinopathy. Exp Eye Res 1999; 68: 91–98.
Alexander JP, Bradley JM, Gabourel JD, Acott TS . Expression of matrix metalloproteinases and inhibitor by human retinal pigment epithelium. Invest Ophthalmol Vis Sci 1990; 31: 2520–2528.
Grant MB, Caballero S, Tarnuzzer RW, Bass KE, Ljubimov AV, Spoerri PE, et al. Matrix metalloproteinase expression in human retinal microvascular cells. Diabetes 1998; 47: 1311–1317.
Das A, McGuire PG, Eriqat C, Ober RR, DeJuan E Jr, Williams GA et al. Human diabetic neovascular membranes contain high levels of urokinase and metalloproteinase enzymes. Invest Ophthalmol Vis Sci 1999; 40: 809–813.
Friedlander M, Theesfeld CL, Sugita M, Fruttiger M, Thomas MA, Chang S et al. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci USA 1996; 93: 9764–9769.
Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA . Definition of two angiogenic pathways by distinct alpha v integrins. Science 1995; 270: 1500–1502.
Amin R, Puklin JE, Frank RN . Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Invest Ophthalmol Vis Sci 1994; 35: 3178–3788.
Hammes HP, Brownlee M, Jonczyk A, Sutter A, Preissner KT . Subcutaneous injection of a cyclic peptide antagonist of vitronectin receptor-type integrins inhibits retinal neovascularization. Nat Med 1996; 2: 529–533.
Verstraeten TC, Chapman C, Hartzer M, Winkler BS, Trese MT, Williams GA . Pharmacologic induction of posterior vitreous detachment in the rabbit. Arch Ophthalmol 1993; 111: 849–854.
Hikichi T, Yanagiya N, Kado M, Akiba J, Yoshida A . Posterior vitreous detachment induced by injection of plasmin and sulfur hexafluoride in the rabbit vitreous. Retina 1999; 19: 55–58.
Gandorfer A, Putz E, Welge-Lussen U, Gruterich M, Ulbig M, Kampik A . Ultrastructure of the vitreoretinal interface following plasmin assisted vitrectomy. Br J Ophthalmol 2001; 85: 6–10.
Trese MT . Enzymatic vitreous surgery. Semin Ophthalmol 2000; 15: 116–121.
Kunisaki M, Bursell SE, Clermont AC et al. Vitamin E prevents diabetes-induced abnormal retinal blood flow via the diacylglycerol-protein kinase C pathway. Am J Physiol 1995; 269: E239–E246.
Keegan A, Walbank H, Cotter MA, Cameron NE . Chronic vitamin E treatment prevents defective endothelium-dependent relaxation in diabetic rat aorta. Diabetologia 1995; 38: 1475–1478.
Bursell SE, Clermont AC, Aiello LP, Aiello LM, Schlossman DK, Feener EP et al. High-dose vitamin E supplementation normalizes retinal blood flow and creatinine clearance in patients with type 1 diabetes. Diabetes Care 1999; 22: 1245–1251.
Duh E, Aiello LP . Vascular endothelial growth factor and diabetes: the agonist versus antagonist paradox. Diabetes 1999; 48: 1899–1906.
Duh E, Aiello LP . Vascular endothelial growth factor and diabetes: the agonist versus antagonist paradox. Diabetes 1999; 48: 1899–1906.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aiello, L., Cahill, M. & Cavallerano, J. Growth factors and protein kinase C inhibitors as novel therapies for the medical management diabetic retinopathy. Eye 18, 117–125 (2004). https://doi.org/10.1038/sj.eye.6700585
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6700585
