
Abstract
Human papillomavirus (HPV) is thought to cause some vulval squamous cell carcinomas (VSCC) by degrading p53 product. Evidence on whether HPV-negative VSCC results from p53 mutation is conflicting. We performed immunohistochemistry for p53 product on 52 cases of lone vulval intraepithelial neoplasia (VIN), 21 cases of VIN with concurrent VSCC and 67 cases of VSCC. We had previously performed HPV detection and loss of heterozygosity (LOH) analyses on these samples. Abnormal p53 immunoreactivity (p53-positive) rates in HPV-positive VSCC and HPV-negative VSCC were 22% (12/54) and 31% (4/13), respectively (P<0.74). p53 immunoreactivity was associated with LOH at the p53 locus (P<0.004), but neither technique differentiated between HPV-positive and HPV-negative VSCC. p53 immunoreactivity was associated with overall LOH rates (p53-positive VSCC vs p53-negative VSCC mean fractional regional allelic loss 0.41 vs 0.24, respectively, P<0.027). LOH at 3p25 was more frequent in p53-positive VSCC cf p53-negative VSCC (70 vs 21%, respectively, P<0.007). There was a trend in p53 disruption associated with invasive disease; HPV-positive VSCC demonstrated more disruption than VIN associated with VSCC, which had more disruption than lone VIN III (22 vs 10 vs 0%, respectively, P<0.005). In all, three out of 73 cases of VIN were p53-positive. All three were associated with concurrent or previous VSCC. Meta-analysis of previous studies revealed significantly more p53 disruption in HPV-negative VSCC cf HPV-positive VSCC (58 vs 33%, respectively; P<0.0001). p53 immunoreactivity/mutation in VIN only appeared in association with VSCC. These data suggest that HPV-independent vulval carcinogenesis does not exclusively require disruption of p53, p53 disruption may work synergistically with LOH at specific loci and p53-positive VIN should be checked carefully for the presence of occult invasion.
Similar content being viewed by others

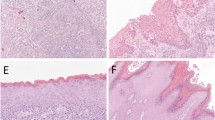
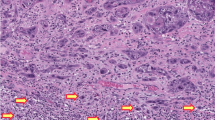
Main
The p53 tumour suppressor gene is central to the development of many solid tumours. It plays key roles in cell cycle regulation and apoptosis. Oncogenic human papillomavirus (HPV) types are thought to cause cervical squamous cell carcinoma. The oncogenic HPV gene products E6 and E7 act on p53 and Rb, respectively. E6 targets p53 product for degradation via the ubiquitin pathway (Scheffner et al, 1990), while E7 complexes and inactivates Rb (Dyson et al, 1989). A proportion of vulval squamous cell carcinoma (VSCC) and the vast majority of vulval intraepithelial neoplasia (VIN) are associated with oncogenic HPV infection (Hording et al, 1994; Kohlberger et al, 1998; Rosenthal et al, 2001). The different patients' ages (Hording et al, 1994; Monk et al, 1995), histological subtypes (Hording et al, 1994; Monk et al, 1995) and the presence of VIN (Hording et al, 1994; Ngan et al, 1999) in HPV-positive but not HPV-negative VSCC, all support the hypothesis that VSCC can arise via both an HPV-dependent and HPV-independent pathway. Few studies have addressed potential differences in the molecular events in these two groups. Loss of heterozygosity (LOH) data from ourselves (Rosenthal et al, 2001) and others (Flowers et al, 1999; Pinto et al, 1999) suggest that HPV-positive and HPV-negative VSCC consistently undergo loss of different chromosomal loci. In addition, another study found differing DNA ploidy in HPV-positive and HPV-negative VSCC (Scurry et al, 1999).
We hypothesised that as HPV-positive VSCC was likely to involve p53 dysfunction because of viral E6 gene product, HPV-negative VSCC might also involve abrogation of the function of this tumour suppressor gene. Data on p53 in this context are conflicting. While some studies suggest that p53 mutation is absent (Kurvinen et al, 1993; Kim et al, 1996) or rare (Lee et al, 1994) in HPV-positive VSCC, but present in about one-third of HPV-negative VSCC, other studies, using a variety of techniques, including immunohistochemistry (IHC), have found 31–48% of HPV-positive VSCC and 58–75% of HPV-negative VSCC to show aberrant p53 expression and/or mutation (Milde-Langosch et al, 1995; Pilotti et al, 1995; Kagie et al, 1997a; Flowers et al, 1999; Ngan et al, 1999). Although all these studies show a trend towards higher proportions of HPV-negative VSCC exhibiting p53 dysfunction compared with HPV-positive samples, none of these studies reached statistical significance. We therefore wanted to try to clarify this issue in a large series and perform a meta-analysis of all these series.
We also wanted to investigate p53 immunoreactivity as a marker of progression to invasion. Some studies suggest absent p53 immunoreactivity in lone VIN (Kurvinen et al, 1993; Tervahauta et al, 1993; Pilotti et al, 1995; Kohlberger et al, 1998), while others have found 17–52% p53 immunoreactivity in VIN associated with VSCC (Milde-Langosch et al, 1995; Kagie et al, 1997a, 1997b; McConnell et al, 1997; Emanuels et al, 1999). It is therefore possible that p53 immunoreactivity could represent a marker for risk of invasion. Higher rates of p53 immunoreactivity in VIN from patients with VSCC compared to those without could indicate a role for this gene in progression from VIN to VSCC. Finally, because we had already investigated our samples for LOH at six different chromosomal loci (Rosenthal et al, 2001), it was possible to examine any relation between aberrant p53 immunoreactivity and LOH at specific loci. Here we report the first study of p53 IHC in a large series of VIN and VSCC samples, also tested for HPV infection and LOH at multiple chromosomal loci.
Materials and methods
Samples
Patients with VIN and VSCC diagnosed between 1989 and 1997 were identified using the computerised database of the pathology departments of St Bartholomew's and the Royal London Hospitals. Samples obtained were as follows: 43 cases of lone VIN III, four cases of lone VIN II, five cases of lone VIN I, 49 cases of VSCC and 21 cases of VIN associated with concurrent VSCC. In all, 18 of the 21 cases of VIN associated with concurrent cancer had the VSCC still remaining on the specimen blocks after serial sectioning. There were therefore 67 cases of VSCC available for study (18 cases associated with concurrent VIN and 49 cases not associated with VIN). Of the 21 cases of VIN associated with VSCC, 18 were VIN III, two were VIN II and one was VIN I. Of the 67 VSCC cases, 24 were stage I, 11 were stage II, nine were stage III, three were stage IV, and in 20 information for accurate staging was not available because specimens were biopsies only. The relevant paraffin-embedded tissue samples were serially sectioned as follows: one 4-μm section was mounted, stained with haematoxylin and eosin, covered and used as a reference slide. One 4-μm section was used for p53 IHC.
LOH analyses and HPV DNA detection and typing
The methods used for LOH analysis and for detection and typing of HPV DNA in the samples in this study have been previously described in detail (Rosenthal et al, 2001). Briefly, DNA was extracted from microdissected archival normal and neoplastic vulval tissue. LOH analysis was performed by PCR using microsatellite markers for the following loci: 17p13–p53, 9p21–p16, 3p25, 4q21, 5p14 and 11p15. HPV DNA was detected by PCR, using highly sensitive consensus genital-type HPV L1 gene primers GP5+ and GP6+ (Kohlberger et al, 1998). PCR products were run on agarose gels, and amplification bands from samples positive for HPV DNA were cut out of the gel, and the DNA extracted and sequenced. The resulting sequences were compared with known HPV types using a BLAST search.
IHC
Samples were dewaxed in three changes of xylene and rehydrated in three changes of methanol (100, 90 and 70%). Following washing in two changes of distilled water, endogenous peroxidase activity was blocked in 3% H2O2 for 10 min. The samples were washed in two changes of Tris-buffered saline (TBS). Nonspecific binding was blocked using 1 : 10 normal goat serum (Dako, Ely, UK) in phosphate-buffered saline and 0.1% bovine serum albumin (PBS–BSA) for 20 min in a humidity chamber. Slides were incubated overnight in 1 : 50 DO7 anti-p53 mouse monoclonal antibody (Dako, Ely, UK) in PBS–BSA. This step was omitted for negative controls. The samples were then incubated for 45 min in 1 : 100 goat anti-mouse secondary biotinylated antibody (Dako, Ely, UK) in PBS–BSA with 10% normal human serum. The samples were washed in two changes of TBS and incubated for 45 min in 1 : 200 streptavidin–biotin complex (Dako, Ely, UK) in TBS. The samples were washed in two changes of TBS. Staining of primary antibody was performed using the chromagen 3-diaminobenzidine tetrahydrochloride (DAB) (180 mg DAB in 270 ml H2O, 30 ml TBS, 1 ml 0.1 M imidazole and 120 μl 30% H2O2). The slides were counterstained using Mayers Haemalum (Merck, Poole, UK), washed in water, placed briefly in 1% acid-alcohol followed by blueing solution (0.5% disodium tetraborate) and dehydrated in two changes of ethanol. The samples were placed briefly in xylene to restore refractive index and mounted using DPX (Merck, Poole, UK). We used a breast cancer sample known to express p53 as a positive control.
Assessment of p53 staining pattern
Samples were scored as negative (<10% of nuclei positive), positive (10–50% of nuclei positive) or highly positive (>50% of nuclei positive) (Kagie et al, 1997b). These percentages were grossly assessed by counting nuclei. All slides were assessed by two observers (ANR, DH) independently.
Meta-analyses of previous studies of p53 in vulval neoplasia
Studies were identified using the search terms vulva, vulval neoplasia, VIN and p53 on PUBMED. References from the publications retrieved were also obtained. Since a wide variety of techniques for assessing p53 dysfunction have been used, and not all studies assessed HPV status, we have made the following assumptions in combining these results for statistical analysis. Firstly, all VIN was assumed to be HPV-positive, irrespective of whether HPV detection was performed. This assumption is justified by our own data (Rosenthal et al, 2001) and that of others (Hording et al, 1995; Kohlberger et al, 1998), which indicates that >90% of VIN is HPV-positive. Secondly, >‘moderate staining’ or >‘10% of nuclei staining positive’ were taken to indicate aberrant p53 expression for IHC results, irrespective of the classification used in the original papers. This was because the majority of papers used this system. Thirdly, in studies using more than one method to evaluate p53 dysfunction, ‘aberrant p53’ refers to the proportion of samples with aberrant p53 by any technique used in the study, that is, samples showing aberrant p53 with one technique, but not all techniques, were still counted as aberrant. While such assumptions may be controversial, they were applied equally to all studies and to both HPV-positive and HPV-negative samples (where HPV testing was used), thus minimising bias. In a rare condition such as VSCC, combining a large number of small series can reveal important trends, which might otherwise be missed.
Statistical analysis
Proportions of samples showing p53 immunoreactivity and LOH were compared using Fisher's exact test or the χ2 test, where appropriate. In order to take into account the differing proportions of noninformative cases in the different sample groups, we calculated the fractional regional allelic loss (FRL) for each sample (Wistube et al, 1997). FRL for each sample=total number of loci undergoing LOH/total number of informative loci. FRL scores for sample groups were compared using the nonparametric Wilcoxon test. Meta-analysis of pooled results from previous published series was performed using the χ2 test. Significance was taken at the 5% level.
Results
The median age of patients with HPV-positive VSCC was 67 years (range 21–94 years) compared with 77 years in the HPV-negative VSCC patients (range 49–91 years). Only four cases (all HPV-positive VSCC) exhibited >50% of nuclei staining positive with DO7. These cases were combined with the samples exhibiting 10–50% of nuclei staining positive with DO7 to provide the ‘p53-positive’ group.
The results of p53 staining in the different VIN and VSCC sample groups are shown in Table 1. Results for LOH at the p53 locus have been published elsewhere (Rosenthal et al, 2001). The combined IHC and LOH results are shown in Table 2. There was no significant difference in the proportions of HPV-positive and HPV-negative VSCC samples demonstrating aberrant p53 by immunoreactivity either alone (Table 1) or in combination with LOH (Table 2). There was a significant trend in increasing aberrant p53 by immunoreactivity either alone (Table 1) or in combination with LOH (Table 2), going from lone VIN to VIN associated with VSCC to HPV-positive VSCC. There was a significant correlation between p53 immunoreactivity and LOH at p53 in VSCC and VIN (Table 3).
p53 immunoreactivity was significantly associated with overall rates of LOH (median FRL 0.40 vs 0.25, P<0.027 in p53-positive VSCC vs p53-negative VSCC). Figure 1 shows the proportions of p53-positive and p53-negative samples undergoing LOH at six different chromosomal loci. LOH at 17p13 (p53 locus), 9p21 (p16 locus) and 3p25 was more common in p53-positive VSCC compared with p53-negative VSCC, but this only reached statistical significance at 17p13 and 3p25 (57 vs 23%, P<0.028 and 70 vs 21%, P<0.007, respectively).

Proportions of p53-positive and p53-negative samples undergoing LOH at six different chromosomal loci. *P<0.028, **P<0.007.
The meta-analyses of previous studies of p53 in VSCC and VIN, along with our own results, are shown in Table 4 and , respectively. Overall, a significantly higher proportion of HPV-negative VSCC samples demonstrated aberrant p53 by one or more methods used, compared with HPV-positive VSCC samples (Table 4, 33 vs 58%, respectively, P<0.0001). Overall, there was a significantly higher proportion of aberrant p53 in VIN associated with VSCC compared with lone VIN (Table 5, 25 vs 7%, respectively, P<0.0004).
Discussion
We set out to compare the rates of p53 disruption in VIN and HPV-positive and HPV-negative VSCC in an attempt to establish whether p53 might be involved in progression from VIN to VSCC, and whether vulval carcinogenesis in the absence of HPV infection requires disruption of p53. p53 has been extensively studied in vulval carcinogenesis. We are aware of 11 studies that have examined p53 disruption in HPV-typed vulval neoplasia (Kurvinen et al, 1993; Tervahauta et al, 1993; Lee et al, 1994; Milde-Langosch et al, 1995; Pilotti et al, 1995; Kim et al, 1996; Kagie et al, 1997a, 1997b; Kohlberger et al, 1998; Flowers et al, 1999; Ngan et al, 1999). A further four studies (McConnell et al, 1997; Sliutz et al, 1997; Emanuels et al, 1999; Scheistrøen et al, 1999) failed to examine HPV status, but two of these included VIN samples (McConnell et al, 1997; Emanuels et al, 1999). Combining the data from these studies and our own, it appears that VIN associated with VSCC is significantly more likely to demonstrate aberrant p53 function than lone VIN (Table 5, 25 vs 7%, P<0.0004), suggesting that p53 could be a marker to identify women at risk of progression from VIN to VSCC. In addition, p53 immunoreactivity is only ever present in VIN when it is associated with cancer. Even the single case of lone VIN I, which demonstrated abnormal p53 immunoreactivity in our study, came from a women who had invasive VSCC 4 years previously. However, only approximately 25% of VIN associated with VSCC demonstrated aberrant p53 function in the meta-analysis. While this proportion is too low to predict which women with VIN will develop VSCC, it does suggest that any women with VIN staining positive for p53 should be carefully assessed to exclude invasion.
In our series, aberrant p53 (by IHC or LOH and IHC combined) was more frequent in HPV-positive VSCC than in the associated VIN, which in turn had more aberrant p53 than lone VIN, suggesting that p53 might be involved in progression to invasive disease. This is surprising when one considers the probable aetiology of VIN. In cervical neoplasia, HPV E6 and E7 oncoproteins inhibit cell cycle arrest and apoptosis, allowing the proliferation of mutant clones. The almost universal presence of oncogenic HPV types in VIN and VIN-associated VSCC (Beckmann et al, 1991; Hording et al, 1995; Van Beurden et al, 1995; Rosenthal et al, 2001), along with the fact that HPV infection is associated with transcription of E6 and E7 in VIN III (Higgins et al, 1991; Jochmus et al, 1993; Van Beurden et al, 1995) and VSCC (Higgins et al, 1991; Park et al, 1991), suggests that HPV can also induce some cases of VSCC in a manner analogous to its effects in the cervix. If oncogenic HPV precipitates the accumulation of mutations throughout the genome in vulval neoplasia, then p53 mutations may occur. As mutant p53 has a longer half-life than wild type, allowing its recognition by the DO7 antibody, it may be that p53 immunoreactivity in VIN and VSCC may simply be an index of the mutation burden of the tissue. This would also explain our finding of nearly double the FRL in p53-positive VSCC compared with p53-negative VSCC (median FRL 0.40 vs 0.25, respectively, P<0.027).
We found a significant association between LOH at p53 and p53 immunoreactivity (Table 3, P<0.004). However, despite this correlation, LOH at p53 frequently occurred in the absence of p53 immunoreactivity. This is not surprising, as LOH does not prove that the retained allele is mutated. p53-positive VSCC underwent LOH at 3p25 significantly more often than p53-negative VSCC (Figure 1, 70 vs 21%, P<0.007), suggesting that LOH at this locus might act synergistically with p53 dysfunction in the carcinogenic pathway. Interestingly, the Fanconi anaemia complementation group D gene has recently been localised to 3p25.3 (Hejna et al, 2000) and VSCC has been reported in patients with Fanconi anaemia at ages much younger than is typical of VSCC (Arnold et al, 1980; Kennedy and Hart, 1982; Wilkinson et al, 1984).
Meta-analysis reveals that HPV-negative VSCC appears to undergo a significantly increased frequency of p53 disruption compared with HPV-positive VSCC (Table 4, 58 vs 33%, P<0.0001), indicating that p53 is involved in the majority of HPV-negative VSCC. However, our own study and meta-analysis support the view that p53 disruption is not obligatory in HPV-independent pathways of vulval carcinogenesis.
In conclusion, we found that p53 immunoreactivity in VIN is associated with the presence of invasive disease, implying that any case of VIN staining positive for p53 should be checked carefully for the presence of occult invasion. Meta-analysis suggests that HPV-negative VSCC has a significantly greater proportion of aberrant p53 compared to HPV-positive VSCC; however, nearly half of all HPV-negative VSCC has no evidence of p53 dysfunction, thus implicating other molecular events in this pathway. We observed a significant association between p53 immunoreactivity and LOH at 3p25 suggesting that a tumour suppressor at this locus might act synergistically with p53 dysfunction. p53 immuno-reactivity was also significantly associated with FRL, implicating it as an index of the mutation burden of the tissue.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arnold WJ, King CR, Magrina J, Masterson BJ (1980) Squamous cell carcinoma of the vulva and Fanconi anemia. Int J Gynaecol Obstet 18: 395–397
Beckmann AM, Acker R, Christiansen AE, Sherman KJ (1991) Human papillomavirus infection in women with multicentric squamous cell neoplasia. Am J Obstet Gynecol 165: 1431–1437
Dyson N, Howley PM, Munger K, Harlow E (1989) The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243: 934–937
Emanuels AG, Koudstaal J, Burger MP, Hollema H (1999) In squamous cell carcinoma of the vulva, overexpression of p53 is a late event and neither p53 nor mdm2 expression is a useful marker to predict lymph node metastases. Br J Cancer 80: 38–43
Flowers LC, Wistuba II, Scurry J, Muller CY, Ashfaq R, Miller DS, Minna JD, Gazdar AF (1999) Genetic changes during the multistage pathogenesis of human papillomavirus positive and negative vulvar carcinomas. J Soc Gynecol Invest 6: 213–221
Hejna JA, Timmers CD, Reifsteck C, Bruun DA, Lucas LW, Jakobs PM, Toth-Fejel S, Unsworth N, Clemens SL, Garcia DK, Naylor SL, Thayer MJ, Olson SB, Grompe M, Moses RE (2000) Localization of the Fanconi anemia complementation group D gene to a 200-kb region on chromosome 3p25.3. Am J Hum Genet 66: 1540–1551
Higgins GD, Uzelin DM, Phillips GE, Burrell CJ (1991) Presence and distribution of human papillomavirus sense and antisense RNA transcripts in genital cancers. J Gen Virol 72: 885–895
Hording U, Junge J, Daugaard S, Lundvall F, Poulsen H, Bock JE (1994) Vulvar squamous cell carcinoma and papillomaviruses: indications for two different etiologies. Gynecol Oncol 52: 241–246
Hording U, Junge J, Poulsen H, Lundvall F (1995) Vulvar intraepithelial neoplasia III: a viral disease of undetermined progressive potential. Gynecol Oncol 56: 276–279
Jochmus I, Durst M, Reid R, Altmann A, Bijward KE, Gissmann L, Jenson AB (1993) Major histocompatability complex and human papillomavirus type 16 E7 expression in high-grade vulvar lesions. Hum Pathol 24: 519–524
Kagie MJ, Kenter GG, Tollenaar RAEM, Hermans J, Trimbos JB, Fleuren GJ (1997a) p53 Protein overexpression, a frequent observation in squamous cell carcinoma of the vulva and in various synchronous vulvar epithelia, has no value as a prognostic parameter. Int J Gynecol Pathol 16: 124–130
Kagie M, Kenter GG, Tolenaar RAEM, Hermans J, Trimbos JB, Fleuren GJ (1997b) p53 Overexpression is common and independent of human papillomavirus infection in squamous cell carcinoma of the vulva. Cancer 80: 1228–1233
Kennedy AW, Hart WR (1982) Multiple squamous-cell carcinomas in Fanconi's anemia. Cancer 50: 811–814
Kim Y-T, Thomas NF, Kessis TD, Wilkinson EJ, Hedrick L, Cho KR (1996) p53 Mutations and clonality in vulvar carcinomas and squamous hyperplasias: evidence suggesting that squamous hyperplasias do not serve as direct precursors of human papillomavirus negative carcinomas. Hum Pathol 27: 389–395
Kohlberger P, Kainz C, Breitenecker G, Gitsch G, Sliutz G, Kolbl H, Tschachler E, Reinthaller A (1998) Absence of p53 protein overexpression in precancerous lesions of the vulva. Cancer 82: 323–327
Kurvinen K, Tervahauta A, Syrjanen S, Change F, Syrjanen K (1993) The state of the p53 gene in human papillomavirus (HPV)-positive and HPV-negative genital precancer lesions and carcinomas as determined by single-strand conformation polymorphism analysis and sequencing. Anticancer Res 14: 177–182
Lee YY, Wilczynski SP, Chumakov A, Chih D, Koeffler HP (1994) Carcinoma of the vulva: HPV and p53 mutations. Oncogene 9: 1655–1659
McConnell D, Miller ID, Parkin DE, Murray GI (1997) p-53 Protein expression in a population-based series of primary vulval squamous cell carcinoma and immediate adjacent field-change. Gynecol Oncol 67: 248–254
Milde-Langosch K, Albrecht K, Joram S, Schlechte H, Giessing M, Loning T (1995) Presence and persistence of HPV infection and p53 mutation in cancer of the cervix uteri and the vulva. Int J Cancer 63: 639–645
Monk B, Burger RA, Lin F, Parkham G, Vasilev SA, Wilczynski SP (1995) Prognostic significance of human papillomavirus DNA in vulvar carcinoma. Obstet Gynecol 85: 709–715
Ngan HY, Cheung AN, Liu SS, Yip PS, Tsao SW (1999) Abnormal expression or mutation of TP53 and HPV in vulvar cancer. Eur J Cancer 35: 481–484
Park JS, Rader JS, Wu TC, Laimins LA, Currie JL, Kurman RJ, Shah KV (1991) HPV-16 viral transcripts in vulvar neoplasia: preliminary studies. Gynecol Oncol 42: 250–255
Pilotti S, D'Amato L, della Torre G, Donghi R, Longoni A, Giarola M, Sampietro G, de Palo G, Pierotti MA, Rilke F (1995) Papillomavirus, p53 alteration, and primary carcinoma of the vulva. Diagn Mol Pathol 4: 239–248
Pinto AP, Lin M-C, Mutter GL, Sun D, Villa LL, Crum CP (1999) Allelic loss in human papillomavirus-positive and -negative vulvar squamous cell carcinomas. Am J Pathol 154: 1009–1015
Rosenthal AN, Ryan A, Hopster D, Surentheran T, Jacobs IJ (2001) High frequency of loss of heterozygosity in vulval intraepithelial neoplasia (VIN) is associated with invasive vulval squamous cell carcinoma (VSCC). Int J Cancer 94: 896–900
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM (1990) The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63: 1129–1136
Scheistrøen M, Tropé C, Pettersen EO, Nesland JM (1999) p53 Protein expression in squamous cell carcinoma of the vulva. Cancer 85: 1133–1138
Scurry J, Hung J, Flowers L, Kneafsy P, Gazdar A (1999) Ploidy in human papillomavirus positive and negative vulvar squamous cell carcinomas and adjacent skin lesions. Int J Gynecol Cancer 9: 187–193
Sliutz G, Schmidt W, Tempfer C, Speiser P, Gitsch G, Elder S, Schneeberger C, Kainz C, Zeillinger R (1997) Detection of p53 point mutations in primary human vulvar cancer by PCR and temperature gradient gel electrophoresis. Gynecol Oncol 64: 93–98
Tervahauta AI, Syrjanen SM, Vayrnen M, Saastamoinen J, Syrjanen KJ (1993) Expression of p53 protein related to the presence of human papillomavirus (HPV) DNA in genital carcinomas and precancer lesions. Anticancer Res 13: 1107–1112
Van Beurden M, ten Kate FJW, Smits HL, Berkhout RJM, de Craen AJM, der Vange N, Lammes FB, ter Schegget J (1995) Multifocal vulvar intraepithelial neoplasia grade III and multicentric lower genital tract neoplasia is associated with transcriptionally active human papillomavirus. Cancer 75: 2879–2884
Wilkinson EJ, Morgan LS, Friedrich Jr EG (1984) Association of Fanconi's anemia and squamous-cell carcinoma of the lower female genital tract with condyloma acuminatum. A report of two cases. J Reprod Med 29: 447–453
Wistube II, Lam S, Behrens C, Virmani AK, Fong KM, LeRiche J, Samet JM, Srivastava S, Minna JD, Gazdar AF (1997) Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst 89: 1366–1373
Acknowledgements
We thank Mr Alex Brown and his staff for cutting and mounting the tissue slides, Mr Steve Jones for providing the list of patients with VIN and VSCC, Ms Susanne Jordan for assisting us with the p53 immunohistochemistry, and Ms Janice Thomas, Dr Philippe van Trappen and Mr Mauro Santibanez-Koref for assisting us with the statistical analyses. We are grateful to Ms Kirsten Rolfe, Dr Chris Perret and Prof. Alan MacLean for their advice and the use of their immunohistochemistry laboratory. ANR was supported by the Gynaecology Cancer Research Fund, AR was supported by the Stroyberg Vagn Jensen foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Rosenthal, A., Hopster, D., Ryan, A. et al. Immunohistochemical analysis of p53 in vulval intraepithelial neoplasia and vulval squamous cell carcinoma. Br J Cancer 88, 251–256 (2003). https://doi.org/10.1038/sj.bjc.6600677
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6600677
