
Abstract
Ten years ago, the first finding of apoptotic cell death on the ‘crime scene’ of cardiac ischemia/reperfusion injury profoundly dismayed the scientific community. This observation jarred with the deeply rooted conviction that cardiac myocytes stoically ‘break, but do not bend’ in the fight against ischemia, instead of spontaneously accepting a peaceful demise for the greater good. Ten years later, a number of studies not only proved right the coexistence of necrosis and apoptosis on the ischemic battle field, but also implicated myocyte apoptosis in the pathogenesis of all the shapes and shades that cardiac ischemic injury can take on.
Similar content being viewed by others
Introduction
Sudden rupture of a vulnerable atherosclerotic plaque in a coronary artery leads to coronary occlusion with resulting acute myocardial infarction (AMI). In this event, myocardial injury ensues as a consequence of the imbalance between oxygen supply and demand. The estimated annual incidence from AMI is 865 000 (including 565 000 new and 300 000 recurrent AMI), with a mortality rate of 184 757 in 2001.1 Reperfusion therapy aimed at inducing pharmacological and/or invasive reopening of the occluded coronary artery was shown to reduce the extent of infarctions, as well as to improve survival, provided that it was started as soon as possible after the onset of ischemia.2 However, if coronary flow is not re-established or is re-established with delay, postischemic cardiac cell loss and ventricular remodeling progress to heart failure in up to 40% of patients.3 Prevention of postischemic cell death still appears to hold the most promise, although there is hope that cardiac repair eventually may be accomplished by cell replacement either through induction of cell division4 or replacement by stem cells.5
Periods of ischemia longer than 20 min inevitably result in irreversible cardiac damage with subsequent cell loss by necrosis and apoptosis. Apoptosis is a highly controlled cell death process, which is autonomously committed by fully healthy or sublethally injured cells in response to physiological or pathological stimuli. This suicidal scheme is achieved through a complex series of ordered biochemical events and finalized without any inflammatory response.6 In contrast to apoptosis, necrosis commonly arises from fatal external insults and results in spillage of the cellular content, with subsequent inflammation.7 Unlike necrotic cell death, which is unanimously recognized as the prototype of ischemic cell death, clinically diagnosed by documentation of myocyte release of cytosolic constituents (such as creatine kinase MB, troponins and other proteins),8 apoptosis has been implicated in the pathogenesis of several acute and chronic conditions affecting the cardiovascular system only in the last decade.9 The involvement of apoptosis in the pathogenesis of cardiovascular diseases was discounted, since apoptosis was initially assumed to be restricted to dividing cells developing tissues and homeostasis of continuously renewing cell populations.10 This view has been amended to recognize that terminally differentiated, nondividing cells can undergo apoptosis in response to pathologic insults such as ischemia/reperfusion.
Apoptosis in the ischemic and reperfused myocardium
Myocardial ischemia occurs when the oxygen demand of the heart exceeds the blood supply, as a consequence of a coronary obstruction.11 Postischemic reperfusion follows resumption of the flow through the vascular bed furnishing the ischemia area. The apoptotic process has been seen to affect cardiac myocytes of ischemic/reperfused hearts, occurring primarily during reperfusion. Gottlieb, who first evaluated the occurrence of apoptosis in an animal model of myocardial ischemia/reperfusion injury, detected myocyte and endothelial cell apoptosis in ischemic/reperfused rabbit hearts exposed to 30 min of ischemia followed by 4 h of reperfusion, although not in normal or continuously ischemic myocardium.12 Loss of endothelial cells precedes and may predispose to cardiomyocyte apoptosis,13 indicating that salvaging endothelial cells is of paramount importance. Whether myocyte apoptosis was initiated during ischemia but dependent on reperfusion or whether it was a feature of reperfusion injury awaited further study. Fliss and Gattinger14 documented that reperfusion accelerated the appearance of apoptosis in rat hearts subjected to 45 min of ischemia followed by 60 min of reperfusion, but reduced the number of apoptotic cells when compared to continuously ischemic tissue, in which the development of apoptosis was slower but more extensive. Freude reported apoptosis after 6 h of reperfusion, among a percentage of necrotic cells higher than 90%, in dog hearts exposed to global ischemia for 90 min.15
The temporal distribution of apoptotic cell death in the two distinct phases of the process was further investigated in order to clarify not only the relative contribution of ischemia alone in triggering apoptosis but also the role played by reperfusion injury either in inducing apoptosis by itself and/or in completing the apoptotic process already commenced during ischemia. This task proved to be anything but easy. Part of the problem was methodological and related to the inability of the techniques at our disposal to distinguish both decisively and in a time-resolved manner between apoptosis and necrosis. The most commonly used technique to detect apoptosis in tissue sections is TUNEL staining, an enzymatic reaction that labels with modified nucleotides the free 3′-hydroxy ends of fragmented DNA, characteristic of the apoptotic process.16 However, since DNA fragmentation is a relatively late event in the apoptotic process, this does not exclude the possibility that some cardiac myocytes from ischemic/reperfused hearts have already initiated the apoptotic program, although undetectable by TUNEL assay. In order to reduce the possibility of under-rating the incidence of cardiac apoptosis and misreading the time course of the process in the acute situation, the data supplied by TUNEL have been integrated with additional techniques, enabling molecular identification of changes occurring during apoptosis earlier than DNA fragmentation. With respect to apoptotic molecular hallmarks, caspase activation is one mechanism widely implicated in the initiation as well as execution of the process, despite the diversity of signals that can induce apoptotic cell death.17 Detection of cleavage of caspase-3 after ischemia alone in cardiac cells, which are otherwise TUNEL negative,18 and colocalization of cleaved caspase-3 labeling with TUNEL staining during reperfusion,13 suggested that ischemia per se is sufficient to initiate apoptosis, although reperfusion is required to complete the process to the stage of DNA fragmentation.
The role of caspases in myocardial cell death after ischemia/reperfusion is controversial. While a number of studies have documented cardiomyocyte salvage by perfusion of caspase inhibitors,19, 20 a role for calpains in the process has also been demonstrated.21, 22, 23 The existence of redundant systems to ensure degradation of proteins and DNA in the lethally injured cell begs the question why enzymatic destruction is desirable. It has been suggested that apoptotic death does not provoke an inflammatory response, which may have consequences for wound healing and remodeling, a topic we will address later in this review.
Controversy over assessment of cell death
Establishing the relative contributions of apoptosis and necrosis to myocardial ischemia and reperfusion injury is important because of the therapeutic implications. However, since stressors, experimental context, as well as operational definition of apoptosis may greatly differ among investigators, a homogeneous interpretation of the data coming from different studies is inevitably challenging. First of all, many of the foregoing experiments were carried out on rabbits, rats and dogs, yet it may not be reasonable to assume that events follow the same course in different species. It is also an excessive approximation to assume that one can generalize from neonatal to adult cardiac myocytes (which differ in their glycolytic capacity, calcium handling and susceptibility to apoptosis); from cultured cells to the intact heart (which differ in the presence of diverse cell populations and cell–cell connections); or from ex vivo to in vivo studies (which differ in cardiac work, as well as in the concurrence of host immune responses, platelets and other coagulation factors).
Furthermore, the accurate description of the incidence of a phenomenon depends on the duration of the event under study. Apoptotic changes occur in a predictable sequence of variable duration. Suzuki has recently shown that in primary cultures of adult rat cardiomyocytes exposed to H2O2, the entire apoptotic sequence, from stimulation to DNA fragmentation, needs 14 h to be finalized.24 Work by Goldstein showed that although a variable time may elapse after an insult, during which time the cell may appear normal, the apoptotic process, once initiated, proceeds rapidly (the interval from onset of cytochrome c release to phosphatidylserine externalization was less than 2 h).25 Thus, studies that score apoptosis at less-frequent intervals will underestimate the incidence of apoptosis. Underestimation can also occur during the ‘decision phase’, when cells appear morphologically normal.
Apoptosis affecting cardiac myocytes exposed to ischemia/reperfusion injury might require more time than in other experimental settings. In fact, the shortage of ATP caused by ischemia, according to its grade of severity, seems to be pivotal in establishing the type of cell death, which injured cells are fated to undergo.26 In this regard, necrosis would take place when energy supplies are near exhaustion; conversely, a less severe depletion in ATP would be sufficient to start the apoptotic machinery but not to conclude the process, whose culmination would be permitted only after reoxygenation of ischemic myocytes.27 Supporting this observation was the finding that caspase-9 activation elicited by mitochondrial damage cannot take place in the absence of ATP.28
Another possible cause of underestimation of apoptotic cell death in tissue sections appears to be the inaccurate quantification of positive cells by means of techniques based solely on the detection of DNA cleavage. In line with this interpretation, Takemura documented that TUNEL-negative myocytes could otherwise exhibit morphological features indicative of apoptosis.29 In contrast, other reports have sternly criticized the specificity of TUNEL, both postulating that TUNEL-positive myocytes may also be oncotic,30 and showing occurrence of DNA replication in cells labeled by TUNEL staining.31 In addition, a study from Kahno reported that cells undergoing DNA repair might also show TUNEL positivity.32 In spite of the sharp censure, a more extensive rendering of the literature seems to indicate that TUNEL technique has survived the line of criticism. It has been shown that TUNEL-positive cells are not seen in normal hearts and are distinct from the unstained necrotic areas produced by prolonged ischemia.33 Moreover, TUNEL accuracy seems to be further validated by the recent introduction of molecular probes regarded as being the most decisive method of differentiating apoptosis from necrosis, since they are capable of recognizing different aspects of DNA fragmentation.34 In fact, when TUNEL reliability was tested by assessing apoptosis in the myocardium with PCR-generated Taq polymerase probes, both techniques produced similar results.35, 36 Furthermore, other in vivo experiments supporting the employment of TdT have provided evidence that necrotic myocytes specifically stained by anti-myosin antibody are TUNEL negative.37
Mitochondrial dysfunction in ischemia/reperfusion
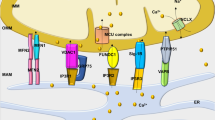
Programmed cell death in the heart shares common features with the process taking place in other cell types. Both receptor-dependent signaling and the mitochondrial pathway have been shown to contribute to cell death.38 Given the critical role of the mitochondria in meeting the energy requirements of the cell, it is clear that preserving mitochondrial viability is essential to preserving overall cardiac function. Mitochondria provide ATP, and participate in calcium homeostasis in the normal setting. In the context of ischemia, mitochondria represent a potent source of reactive oxygen species (ROS), may serve as an additional drain on limited energy stores (if the F0F1 ATPase is allowed to run in reverse) and can release cytochrome c, an essential cofactor for caspase-9 activation. In addition, ischemic mitochondria, which initially sequester calcium in a futile and energy-consuming cycle, may eventually release it, along with cytochrome c and other proapoptotic factors, in a catastrophic bolus, upon opening of the mitochondrial permeability transition pore.39 Recent work has shown that ischemic or pharmacologic preconditioning raises the threshold for mitochondrial permeability transition pore opening, underscoring the importance of preserving mitochondrial integrity.40, 41 Loss of homeostatic ability, failure of ATP production, excessive ROS and activation of apoptosis all conspire to ensure the destruction of the cell, whose mitochondria are irretrievably damaged. These competing processes may result in cell death that resembles either apoptosis or necrosis. Interventions that enable preservation of mitochondrial integrity will attenuate both forms of cell death. These include, among the others, overexpression of antiapoptotic Bcl-2 family members,42 overexpression or protein transduction of the apoptosis repressor with caspase recruitment domain (ARC),43 induction and opening of mitochondrial K(ATP) channel,44 as well as oral supplementation with mixed essential amino acid.45
Apoptosis and persistent severe ischemia
Translocation of phosphatidylserine from the inner to the outer leaflet of the cell membrane is a well-recognized immunological signal by means of which apoptotic cells and apoptotic bodies are engulfed by competent phagocytic cells provided with specific phosphatidylserine receptors.46, 47 However, although apoptosis is effectively distinguished from necrosis by the retention of intracellular contents, delayed or impaired removal of apoptotic cells by phagocytes inevitably results in secondary necrosis of the apoptotic remnants over a period of hours. This shift is most likely to take place in cardiac cells exposed to continuous severe ischemia. Evidence of the transition from apoptotic to necrotic cell death in the heart was first provided by Kajstura, who evaluated quantitatively from 20 min to 7 days after permanent coronary occlusion the relative contribution of apoptosis and necrosis to infarct size in a rat model of myocardial infarction.48, 49 After 2 h of persistent ischemia, in the infarcted portion of the left ventricle 2.7 millions of cardiac cells were apoptotic and only 90 000 necrotic. The magnitude of apoptotic cell death peaked at 4.5 h, while necrosis reached a maximum level at 1 day after coronary ligation. Although apoptotic cell death was shown to be the major independent variable contributing to infarct size, accounting for 86% of total cell loss, the authors also emphasized that apoptotic myocytes in the infarcted myocardium acquired necrotic features, with heavy switch from apoptosis to necrosis in the late stages of ischemia. A transition phase characterized by findings of cardiac cells showing concurrently the stigmata of both apoptotic and necrotic cell death was also documented in human autopsied cardiac tissue from cases of AMI.50, 51, 52 Evidence of apoptotic cell death was found not only in the core of fresh myocardial infarctions but also in both the hypoperfused border zone (between central infarct area and surviving myocardial tissue) and the remote noninfarcted myocardium.53, 54 Quantitative assessment of apoptotic cell death documented TUNEL-positive staining in 12% of myocytes in the border zone, whereas 1% of cardiac cells were undergoing apoptosis in the remote myocardium, suggesting that apoptosis was a complicating factor of AMI, leading to extension of total myocyte cell death following an acute coronary occlusion.53 Data from animal models of ischemia/reperfusion injury seem to indicate that the occurrence of apoptotic cell death in the surviving portion of the ischemic ventricle mainly depends on the duration of the ischemic insult and the extent of the resultant infarct size. Multiple infarctions due to embolism of small coronary arteries induced by microsphere injection in dogs55 and very large infarctions induced by permanent coronary occlusion in rats,56 both resulting in severe heart failure, have been associated with the presence of TUNEL-positive myocytes in the surviving tissue surrounding the old infarction. Conversely, in the rabbit heart reperfused after exposure to 30 min of ischemia, which resulted in a relatively small infarct size with no evidence of heart failure, TUNEL-positive staining in salvaged myocytes was very rare, suggesting that the discrepant results between these studies would be resolved by the adoption of different animal models of ischemia/reperfusion injury, with and without the complication of postischemic heart failure.57
Apoptosis in the healing heart and ventricular remodeling following myocardial infarction
During the acute inflammatory phase following a myocardial infarction, the necrotic area is infiltrated by a large population of inflammatory cells, including neutrophils and macrophages. These cells are progressively replaced by granulation tissue, which consists of a variegated cellular pool, comprising myofibroblasts, macrophages and endothelial cells, and finally evolves to the formation of a permanent cardiac scar. The progression of the histological events, which lead to healing of the heart after an AMI, have been characterized and reported both in animals and humans.58, 59 Takemura documented that apoptotic cell death contributes importantly to the removal of the various cell populations, which inhabit the damaged myocardium during the acute and subacute phase of the repair process.57 However, although endothelial apoptosis seems to account for the progressive loss of vessels and reduced blood supply to the healing area, playing therefore a straightforward negative role, it is still controversial whether myofibroblast apoptosis would be beneficial or detrimental. Myofibroblasts, which have been identified in both rat60 and man, are interstitial spindle-shaped cells59 sharing features of fibroblast and smooth muscle cells.61 In addition to collagen synthesis,61 which seems to be their predominant functional role, it was suggested that myofibroblasts would provide the wound with contractile properties.62 However, although the massive appearance and proliferation of myofibroblasts in the infarcted area followed by their progressive demise by apoptosis would imply a role for myofibroblasts in the pathogenesis of abnormal ventricular remodeling, the factual contribution of myofibroblasts to healing and remodeling of the infarcted ventricle is still unknown. Since myofibroblasts in the infarcted area may contribute to the scar formation by collagen synthesis, it follows that promotion of myofibroblast apoptosis may result either in attenuation of pathological fibrosis in remodeling or in structural penchant to aneurismal development due to diminished connective tissue. Conversely, lessening of myofibroblast apoptosis may either reduce the risk of ventricular dilation, making scar tissue collagen-rich, or achieve the opposite effect of causing prolongation of fragile granulation tissue.
Subsequent studies, investigating the causal role of myocyte apoptosis in the pathogenesis of left ventricle remodeling following myocardial infarction, correlated increased apoptotic rates with specific features of postinfarct remodeling. In the rat heart surgically exposed to permanent coronary occlusion, Palojoki documented a correlation between diastolic left ventricular diameters (1–4 weeks after infarction) and cardiomyocyte apoptosis in the remote noninfarcted myocardium, although not in the border zones.63 Furthermore, in a post-mortem study conducted on a small population of patients who died within 10 to 62 days after a myocardial infarction, accelerated myocyte apoptosis in the infarcted and remote areas correlated with left ventricular dilation and thinning of noninfarcted remote myocardium, respectively.64
Myocyte apoptosis and iatrogenic ischemia/reperfusion injury
During cardiopulmonary bypass, the cardioplegic arrest and subsequent reperfusion inevitably expose the human heart to an iatrogenic ischemia/reperfusion injury. Although different cardioplegic techniques (crystalloid, cold and warm blood cardioplegia) have been developed to prevent this injury,65 the protection given to the heart by cardioplegia is often inadequate, especially for surgical procedures requiring prolonged cardiac arrest. In a study carried out in patients receiving three different cold antegrade crystalloid cardioplegic solutions (St. Thomas II, Bretschneider or Hamburg, Germany), DNA fragmentation assessed by TUNEL staining in atrial biopsies was qualitatively documented in subendocardial myocytes and endothelial cells following cardioplegic arrest and reperfusion, although not in prebypass samples.66 Likewise, functional and ultrastructural changes occurring in cardiac myocytes and the coronary circulation have been described in the human heart protected by cold blood cardioplegia.67, 68 More recently, also warm blood cardioplegia, which nowadays is regarded as the state of art of intraoperative myocardial protection,69, 70 has been associated with myocyte apoptosis. In a quantitative study evaluating the occurrence of apoptosis in atrial myocytes from patients exposed to cardiopulmonary bypass, warm blood cardioplegia and subsequent reperfusion, >3% of cardiac cells showed colocalization of TUNEL and cleaved caspase-3 staining after 40–55 min of cardioplegic arrest followed by 10 min of reperfusion, while the proportion of positive myocytes was almost doubled in patients exposed to roughly twice the duration of cardioplegic arrest followed by the same reperfusion time.71 This study confirmed that, in patients undergoing on-pump cardiac surgery, cardioplegia may ameliorate, but does not totally prevent, myocyte apoptosis, whose rates significantly correlate with the length of time on bypass. The loss and functional impairment of myocytes following cardioplegic arrest are known to reduce cardiac contractility directly, resulting in greater mortality and morbidity associated with on-pump cardiac surgery.67, 68 Although atrial changes may not fully reflect cell death in ventricular myocytes, these findings suggest that minimization of myocyte apoptosis may possibly reduce the risk of postsurgical cardiac dysfunction in patients exposed to the inescapable ischemia/reperfusion injury associated with cardiopulmonary bypass surgery.
In vivo visualization of myocyte apoptosis in ischemia/reperfusion injury
As stated previously, apoptotic cells show early redistribution of phosphatidylserine residues from the inner to the outer leaflet of the plasma membrane lipid bilayer.46, 47 This externalization process is a standardized trait of apoptotic cell death occurring earlier than DNA degradation. The human protein annexin V was shown to bind with high affinity to cells exposing phosphatidylserine both in vitro47, 66, 72 and in vivo.73, 74 On the basis of these encouraging results, annexin V, either alone or coupled to technetium 99m, was injected in a rat model of acute cardiac allograft rejection for in vivo evaluation of apoptotic cell death.75 Scintigraphic radionuclide imaging showed in cardiac allografts a net increase in the uptake of radiolabeled annexin V, while antiannexin V immunostaining revealed intense positivity of numerous myocytes, related to the occurrence of both necrotic and apoptotic cell death. These findings suggested that in vivo localization of annexin V is not exclusively specific for apoptosis, given the ability of annexin V to insinuate through the disrupted membranes of necrotic cells, and bind to their internally located phosphatidylserine residues. In a successive study, Dumont used for the first time labeled human recombinant annexin V in order to detect myocyte cell death in an in vivo mouse model of cardiac ischemia/reperfusion injury.76 Annexin V-positive labeling was found in 1.4% of cardiac cells after 15 min of ischemia and 30 min of reperfusion, and this number increased to >11% when the same ischemic period was followed by 90 min of reperfusion. The specific activation of apoptotic cell death was confirmed performing DNA gel electrophoresis, which showed typical laddering starting after 15 min of ischemia followed by 30 min of reperfusion. The binding of fluorescent-labeled annexin V to single cardiomyocytes was subsequently visualized by means of a high-magnification real-time imaging system in the heart of living mice undergoing ischemia/reperfusion injury. In line with previous findings,13 labeling of cardiac myocytes imaged at the single-cell levels was shown to start after a few minutes of reperfusion preceded by 30 min of ischemia, and climaxed after 20–25 min of reperfusion.77 The challenging task to detect apoptotic cell death in vivo in a clinical setting was finally addressed by Hofstra in patients with AMI subjected to primary coronary angioplasty.78 Among seven recruited patients, six of them developed in the early stages of reperfusion increased technetium 99m-labelled annexin V uptake in areas of the myocardium with contextual matching perfusion defects, which suggests occurrence of cell death in the infarcted region, although not in the remote areas. Although qualitative (or at most semiquantitative) and not suitable for the specific detection of apoptosis, this molecular imaging technique holds the unique potential to provide a noninvasive description of the time kinetics of both necrotic and apoptotic cell death in the entire human heart exposed to ischemia reperfusion injury.
Apoptosis and hibernating myocardium
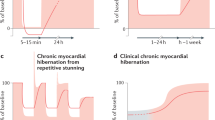
In patients with critical coronary stenoses, the hypoperfused myocardium exposed to chronic or repetitive ischemic insults undergoes a ‘hibernation’ process, which is mainly characterized by dysfunctional contractile performance and preserved cellular viability.79 A functional downregulation of the hibernating heart, thereby allowing the attainment of a metabolic balance between oxygen demand and supply, has been suggested as a feasible explanation of the retained myocardial viability. This prerogative of the dysfunctional myocardium is confirmed by striking pieces of evidence, namely the ability of the hibernating myocardium to provide a contractile response to low-concentration inotropic stimulation,80 the enhanced glucose uptake documented by positron emission tomography (PET) in the hibernating segments81 and the contractile recovery of cardiac function following surgical revascularization procedures.82
In patients with delayed recovery in cardiac function following bypass cardiac surgery, the histological evaluation of cardiac biopsies obtained from the hibernating regions documented the presence of cardiac cells undergoing a dedifferentiation process, with structural and biochemical features of fetal myocytes.83 The additional finding in the same samples of an increased interstitial space in the absence of myocyte atrophy put forward the question of whether cell death could occur. In a pig model of cardiac hibernation, following persistent and severe, although not complete, coronary stenosis, myocyte apoptosis was detected by TUNEL assay, occasionally in association with patchy necrosis.84 Qualitative occurrence of myocyte apoptosis was also observed in the human hibernating myocardium in conjunction with reparative fibrosis, as well as disorganization of contractile and cytoskeletal proteins, suggesting that cellular degeneration, rather than adaptation, is the main histological feature of cardiac hibernation.85 Although lacking a quantitative connotation, these data have important clinical implications. Occurrence of ongoing myocyte apoptosis in the chronic condition of cardiac hibernation, even though at very low rates, in a long-term perspective would certainly result in substantial cardiac cell loss, with time-dependent reduction of the possibility to achieve full structural and functional recovery after adequate restoration of the coronary flow. In line with this speculation, in patients with multivessel coronary disease and myocardial hibernation undergoing coronary bypass surgery, the postoperative recovery in cardiac function was inversely proportional to the severity of the morphological changes86 and enhanced in patients with a short history of hibernation, suggesting that early revascularization should be recommended to minimize the progression of cell degeneration in the hibernating myocardium and improve postoperative outcome.86
Functional repercussions of apoptosis in the heart
The role exerted by myocyte apoptosis in contributing to the extent of cardiac damage following ischemia/reperfusion injury has been extensively investigated. Caspase inhibition by means of broad-spectrum87, 88 and selective cysteine protease inhibitors,38 pharmacological modulation of primary and secondary caspase activation,89 mitochondrial protection achieved by overexpression of the antiapoptotic gene Bcl-2,42, 90 attenuation of the death receptor-mediated pathway by cardiac inhibition of TNF-α91 and the creation of multiple genetic mutations in the apoptotic signaling pathways92 have documented that inhibition of apoptosis in the ischemic/reperfused heart by means of diverse approaches induced not only a reduction in cell death, as expected, but also, more importantly, an enhancement of postischemic recovery of cardiac function. This consideration is undoubtedly of paramount importance since the finding of minimized cell death not associated with enhancement of cardiac function would have only limited translational power and therefore clinical relevance.
Cardiac dysfunction is likely to occur also during the early stages of apoptosis, and independently from the completion of the process. Caspase activation, for instance, has been associated with early myofibrillary protein cleavage, resulting in decreased ATPase activity and contractile dysfunction, before the occurrence of the typical DNA fragmentation detected by TUNEL staining.93 In line with these findings, cardiac broad myofibrillary loss, together with extensive myocyte activation of caspase-9 and -3, largely independent of DNA fragmentation, was also documented in a case of sudden death temporally related to ephedra intake.94 Since caspase activation was also detected during relatively short periods of ischemia alone, in the absence of DNA cleavage,13, 18 it is possible that activated caspases may contribute to cardiac dysfunction also in the early phase of ischemia, inducing precocious breakdown of myofilaments with subsequent contractile impairment, before the completion of the apoptotic process, and independently from necrotic cell death. Considering the massive requirement for mitochondrial ATP production in the heart, it is worth noting that caspase activation can impair mitochondrial respiration.95 However, although captivating and partially supported by experimental and human data, this hypothesis, suggesting a causative role of caspase activation in the induction of cardiac dysfunction, still needs to be validated in the intact heart exposed to ischemia/reperfusion injury.
Increasing the apoptotic/necrotic ratio to modulate the postischemic inflammatory response
In the face of the coexistence in the same pathological context, necrosis and apoptosis are modes of death fully differentiable, not only on the basis of their specific biochemical and morphological features but also in terms of direct repercussions on the affected tissues. Culmination of necrotic cell death with disruption of the cell membrane results in spillage of the cytosolic content with subsequent inflammatory reaction, characterized by leukocyte infiltration and phagocytosis. On the other hand, completion of apoptosis is characterized by formation of several membrane-bound apoptotic bodies, which are engulfed by macrophages and neighboring cells, without a minimal inflammatory response. Although contributing to tissue repair as well as scar formation,96 the inflammatory reaction triggered in the reperfused infarct area can also worsen the ongoing myocardial injury. This has been extensively documented by several studies reporting beneficial effects on cardiac injury and function following attenuation of the inflammatory response associated with myocardial infarction, both in the experimental97, 98, 99, 100, 101, 102 and clinical setting.103, 104 Leukocyte release of proteolytic enzymes in the reperfused infarction, besides promoting disruption of normal myocardial cells and tissue loss within the necrotic zone, can also weaken the connective tissue scaffolding of the heart. When this happens, disproportionate thinning and dilation of the infarct zone prior to the formation of a firm fibrotic scar ensue, leading to a notoriously common complication known as ‘infarct expansion’.105 This complication, which is easily diagnosed by echocardiography as elongation of the infarcted region of the ventricle,106 can be observed in over 75% of patients with myocardial infarction, and is significantly associated with both higher mortality and incidence of nonfatal complications, such as heart failure and ventricular aneurysm.107 More rarely, infarct expansion may result in rupture of the free wall of the infarcted ventricle into the pericardium, leading to progressive and frequently lethal hemodynamic impairment due to cardiac tamponade.108 Therefore, besides the reduction in infarct size, the attenuation of the inflammatory response associated with necrotic cell death also exerts beneficial effects on clinical outcomes in patients with AMI. In this scenario, the inflammation-free removal process of terminal apoptotic remnants would be preferable to necrosis, from the perspective of bystander cells.
In the specific setting of myocardial infarction, the next task will be the development of alternative strategies aimed at enhancing cardioprotection, by modulation of the balance between necrosis and apoptosis within the infarcted area. Endorsement of apoptosis to the detriment of necrosis may lessen postinfarct complications by attenuation of the postischemic inflammatory reaction. Following an infarction, any strategy with the potential to minimize cardiac cell loss should be attempted, including the paradoxical welcome of apoptosis in the infarcted area, as long as it is associated with a reduction in myocyte necrosis. Choosing the less brutal of the two enemies by swapping necrosis with apoptosis may save a number of lives.
Conclusions
The first description of apoptotic cell death in the ischemic/reperfused myocardium provoked consternation, 10 years ago, in the scientific community. This shocking finding dramatically clashed with the prevailing belief that cardiac myocytes stoically ‘break, but do not bend’ in the fight against ischemia, instead of willingly accepting sacrifice for the greater good. After 10 years, a myriad of studies not only proved right the pioneering concept of coexistence of necrosis and apoptosis on the ‘crime scene’ of ischemia/reperfusion injury but also documented that apoptosis is widespread in all the shapes and shades that cardiac ischemic injury can take on. Since the apoptotic death process, unlike the necrotic demise, is started, conducted and finalized through a complex series of ordered biochemical events, specific interventions carried out at different levels may arrest the program, permitting cells to survive and even repair themselves. The capacity of myocytes to self-terminate by means of the energy-dependent mechanism of apoptosis testifies that cells committed to apoptotic demise are still alive and potentially retrievable from the tunnel of death. Myocytes committed to apoptosis are likely to be reversibly damaged, otherwise they would die by necrosis. The notion that a high percentage of cells are salvageable is borne out by variegated interventions, such as ischemic preconditioning,109 urocortin administration,110 protein kinase C modulation,111 cytochrome P450 inhibition112 and Src inhibition,113 which induced substantial reduction in necrotic and apoptotic cell death in experimental models of myocardial ischemia/reperfusion. Some caution must be taken when contemplating applying these encouraging results to humans, since all experimental animal models have been designed to yield informative results by limiting the duration of ischemia, and by utilizing young, healthy and genetically homogeneous animals. It should not be surprising that demonstration of cardiac salvage by any intervention has proven to be considerably more challenging in the clinical setting. Nonetheless, identification of potential targets that can achieve cardiac salvage after ischemia raises hope that reduction of infarct size and postischemic complications is a clinically attainable goal.
Abbreviations
- AMI:
-
acute myocardial infarction
- ROS:
-
reactive oxygen species
- PET:
-
positron emission tomography
References
American Heart Association (2003) Heart Disease and Stroke Statistics – 2004 update. Dallas, Tex.: American Heart Association
Hochman JS and Choo H (1987) Limitation of myocardial infarct expansion by reperfusion independent of myocardial salvage. Circulation 75: 299–306
Sutton MG and Sharpe N (2000) Left ventricular remodelling after myocardial infarction: pathophysiology and therapy. Circulation 101: 2981–2988
Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA and Anversa P (2001) Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl. J. Med. 7;344: 1750–1757
Lee MS and Makkar RR (2004) Stem-cell transplantation in myocardial infarction: a status report. Ann. Intern. Med. 140: 729–737
Searle J, Kerr JFR and Bishop CJ (2000) Necrosis and apoptosis: distinct modes of cell death with fundamentally different significance In Pathology Annual Sommers SC, Rosen PP, eds Norwalk: CT Appleton-Century-Crofts pp. 229–260
Umansky S and Tomei L (2000) Apoptosis in the heart In Apoptosis: Pharmacological Implications and Therapeutic Opportunities Kaufman SH, ed San Diego, CA: Academic Press pp. 383–408
Adams III JE, Schechtman KB, Landt Y, Ladenson JH and Jaffe AS (1994) Comparable detection of acute myocardial infarction by creatine kinase MB isoenzyme and cardiac troponin I. Clin. Chem. 40 (Part 1): 1291–1295
Haunstetter A and Izumo S (1998) Apoptosis: clinical implications for cardiovascular disease. Circ. Res. 82: 1111–1129
Pexieder T (1975) Cell death in the morphogenesis and teratogenesis of the heart. Adv. Anat. Embryol. Cell Biol. 51: 3–99
Jennings RB, Murry CE and Reimer KA (1990) Myocardial effects of brief periods of ischemia followed by reperfusion In Advances in Cardiology Vol. 37, Kellermann JJ, Braunwald E, eds Basel, Switzerland: Karger pp. 7–31
Gottlieb RA, Burleson KO, Kloner RA, Babior BM and Engler RL (1994) Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Invest. 94: 1621–1628
Scarabelli T, Stephanou A, Rayment N, Pasini E, Comini L, Curello S, Ferrari R, Knight R and Latchman D (2001) Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischaemia/reperfusion injury. Circulation 4: 253–256
Fliss H and Gattinger D (1996) Apoptosis in ischemic and reperfused rat myocardium. Circ. Res. 79: 949–956
Freude B, Masters TN, Kostin S, Robicsek F and Schaper J (1996) Cardiomyocyte apoptosis in acute and chronic conditions. J. Mol. Cell. Cardiol. 94: 1621–1628
Bortner CD, Oldenburg NB and Cidlowski JA (1995) The role of DNA fragmentation in apoptosis. Trends Cell Biol. 5: 21–26
Thornberry NA and Lazebnik Y (1998) Caspases: enemies within. Science 281: 1312–1316
Freude B, Masters N, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S and Schaper J (2000) Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J. Mol. Cell. Cardiol. 32: 197–208
Gottlieb RA, Gruol DL, Zhu JY and Engler RL (1996) Preconditioning rabbit cardiomyocytes: role of pH, vacuolar proton ATPase, and apoptosis. J. Clin. Invest. 97: 2391–2398
Holly TA, Drincic A, Byun Y, Nakamura S, Harris K, Klocke FJ and Cryns VL (1999) Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J. Mol. Cell. Cardiol. 31: 1709–1715
Chen M, Won DJ, Krajewski S and Gottlieb RA (2002) Calpain and mitochondria in ischemia/reperfusion injury. J. Biol. Chem. 277: 29181–29186
Chen M, He H, Zhan S, Krajewski S, Reed JC and Gottlieb RA (2001) Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J. Biol. Chem. 276: 30724–30728
Kubasiak LA, Hernandez OM, Bishopric NH and Webster KA (2002) Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc. Natl. Acad. Sci. USA 99: 12825–12830
Suzuki K, Kostin S, Person E, Elsasser A and Shaper J (2001) Time course of the apoptotic cascade and effects of caspase inhibitors in adult rat ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 33: 983–994
Goldstein JC, Waterhouse NJ, Juin P, Evan GI and Green DR (2002) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2: 156–162
Leist M, Single B, Castoldi AF, Kuhnle S and Nicotera P (1997) Intracellular ATP concentration: a switch deciding between apoptosis and necrosis. J. Exp. Med. 185: 1481–1486
Bradbury DA, Simmons TD, Slater KJ and Crouch SP (2000) Measurement of the ADP:ATP ratio in human leukaemic cell lines can be used as an indicator of cell viability, necrosis and apoptosis. J. Immunol. Methods 240: 79–92
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES and Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf1/caspase-9 complex initiates and apoptotic protease cascade. Cell 91: 479–489
Takemura G, Kato S, Aoyama T, Hayakawa Y, Kanoh M, Maruyama R, Arai M, Nishigaki K, Minatoguchi S, Fukuda K, Fujiwara T and Fujiwara H (2001) Characterization of ultrastructure and its relation with DNA fragmentation in Fas-induced apoptosis of cultured cardiac myocytes. J. Pathol. 193: 546–556
Ohno M, Takemura G, Ohno A, Misao J, Hayakawa Y, Minatoguchi S, Fujiwara T and Fujiwara H (1998) ‘Apoptotic’ myocytes in infarct area in rabbit hearts may be oncotic myocytes with DNA fragmentation. Circulation 98: 1422–1430
Kockx MM, Muhring J, Knaapen MW and de Meyer GR (1998) RNA synthesis and splicing interferes with DNA in situ end labeling techniques used to detect apoptosis. Am. J. Pathol. 152: 885–888
Kanoh M, Takemura G, Misao J, Hayakawa Y, Aoyama T, Nishigaki K, Noda T, Fujiwara T, Fukuda K, Minatoguchi S and Fujiwara H (1999) Significance of myocytes with positive DNA in situ nick end-labelling (TUNEL) in hearts with dilated cardiomyopathy. Circulation 99: 2757–2764
Zhao Z, Nakamura M, Wang N, Wilcox JN, Shearer S, Ronson RS, Guyton RA and Vinten-Johansen J (2000) Reperfusion induces myocardial apoptotic cell death. Cardiovasc. Res. 45: 651–660
Buja LM and Entman ML (1998) Modes of myocardial injury and cell death in ischaemic heart diseases. Circulation 98: 1355–1357
Leri A, Claudio PP, Wang X, Reiss K, Wang S, Malhotra A, Kajstura J and Anversa P (1998) Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin–angiotensin system and decreases the Bcl-2 to Bax protein ratio in the cell. J. Clin. Invest. 101: 1326–1342
Li B, Setoguchi M, Wang X, Andreoli AM, Leri A, Malhotra A, Kajstura J and Anversa P (1999) Insulin-like growth factor-1 attenuates the detrimental impact of nonocclusive coronary artery constriction on the heart. Circ. Res. 84: 1007–1019
Anversa P, Leri A, Beltrami CA, Guerra S and Kajstura J (1998) Myocyte death and growth in the failing heart. Lab. Invest. 78: 767–786
Scarabelli TM, Stephanou A, Pasini E, Comini L, Raddino R, Knight RA and Latchman DS (2002) Different signaling pathways induce apoptosis in endothelial cells and cardiac myocytes during ischemia/reperfusion injury. Circ. Res. 5;90: 745–748
Gustafsson AB and Gottlieb RA (2003) Mechanisms of apoptosis in the heart. J. Clin. Immunol. 23: 447–459
Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN and Sollott SJ (2004) Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 113: 1535–1549
Argaud L, Gateau-Roesch O, Chalabreysse L, Gomez L, Loufouat J, Thivolet-Bejui F, Robert D and Ovize M (2004) Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovasc. Res. 61: 115–122
Brocheriou V, Hagege AA, Oubenaissa A, Lambert M, Mallet VO, Duriez M, Wassef M, Kahn A, Menasche P and Gilgenkrantz H (2000) Cardiac functional improvement by a human Bcl-2 transgene in a mouse model of ischemia/reperfusion injury. J. Gene Med. 2: 326–333
Gustafsson AB, Sayen MR, Williams SD, Crow MT and Gottlieb RA (2002) TAT protein transduction into isolated perfusedhearts: TAT-apoptosis repressor with caspase recruitment domain is cardioprotective. Circulation 106: 735–739
Lawrence KM, Chanalaris A, Scarabelli T, Hubank M, Pasini E, Townsend PA, Comini L, Ferrari R, Tinker A, Stephanou A, Knight RA and Latchman DS (2002) K(ATP) channel gene expression is induced by urocortin and mediates its cardioprotective effect. Circulation 106: 1556–1562
Scarabelli TM, Pasini E, Stephanou A, Chen-Scarabelli C, Saravolatz L, Knight RA, Latchman DS and Gardin JM (2004) Nutritional supplementation with mixed essential amino acids enhances myocyte survival, preserving mitochondrial functional capacity during ischemia–reperfusion injury. Am. J. Cardiol. 93: 35A–40A
Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL and Henson PM (1992) Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1;148: 2207–2216
Verhoven B, Schlegel RA and Williamson P (1995) Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J. Exp. Med. 182: 1597–1601
Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, Reed JC, Olivetti G and Anversa P (1996) Apoptotic and necrotic cell death are independent contributing variables of infarct size in rats. Lab. Invest. 74: 86–107
Anversa P, Cheng W, Liu Y, Leri A, Redaelli G and Kajstura J (1998) Apoptosis and myocardial infarction. Basic Res. Cardiol. 93 (Suppl 3): 8–12
Itoh G, Tamura J, Suzuki M, Suzuki Y, Ikeda H, Koike M, Nomura M, Jie T and Ito K (1995) DNA fragmentation of human infarcted myocardial cells demonstrated by the nick end labeling method and DNA agarose gel electrophoresis. Am. J. Pathol. 146: 1325–1331
Veinot JP, Gattinger DA and Fliss H (1997) Early apoptosis in human myocardial infarcts. Hum. Pathol. 28: 485–492
Bardales RH, Hailey LS, Xie SS, Schaefer RF and Hsu SM (1996) In situ apoptosis assay for the detection of early acute myocardial infarction. Am. J. Pathol. 149: 821–829
Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, Gambert SR, Cigola E and Anversa P (1996) Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J. Mol. Cell. Cardiol. 28: 2005–2016
Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M and Voipio-Pulkki LM (1997) Apoptosis in human acute myocardial infarction. Circulation 21;95: 320–323
Sharov VG, Sabbah HN, Shimoyama H, Goussev AV, Lesch M and Goldstein S (1996) Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am. J. Pathol. 148: 141–149
Cheng W, Kajstura J, Nitahara JA, Li B, Reiss K, Liu Y, Clark WA, Krajewski S, Reed JC, Olivetti G and Anversa P (1996) Programmed myocyte cell death affects the viable myocardium after infarction in rats. Exp. Cell Res. 1;226: 316–327
Takemura G, Ohno M, Hayakawa Y, Misao J, Kanoh M, Ohno A, Uno Y, Minatoguchi S, Fujiwara T and Fujiwara H (1998) Role of apoptosis in the disappearance of infiltrated and proliferated interstitial cells after myocardial infarction. Circ. Res. 82: 1130–1138
Fishbein MC, Maclean D and Maroko PR (1978) Experimental myocardial infarction in the rat: qualitative and quantitative changes during pathologic evolution. Am. J. Pathol. 90: 57–70
Willems IE, Havenith MG, De Mey JG and Daemen MJ (1994) The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am. J. Pathol. 145: 868–875
Vracko R, Thorning D and Frederickson RG (1990) Fate of nerve fibers in necrotic, healing, and healed rat myocardium. Lab. Invest. 63: 490–501
Gabbiani G, Ryan GB and Majno G (1971) Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 27: 549–550
Gabbiani G, Hirschel BJ, Ryan GB, Statkov PR and Majno G (1972) Granulation tissue as a contractile organ: a study of structure and function. J. Exp. Med. 135: 719–734
Palojoki E, Saraste A, Eriksson A, Pulkki K, Kallajoki M, Voipio-Pulkki LM and Tikkanen I (2001) Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am. J. Physiol. Heart Circ. Physiol. 280: H2726–H2731
Abbate A, Biondi-Zoccai GG, Bussani R, Dobrina A, Camilot D, Feroce F, Rossiello R, Baldi F, Silvestri F, Biasucci LM and Baldi A (2003) Increased myocardial apoptosis in patients with unfavorable left ventricular remodeling and early symptomatic post-infarction heart failure. J. Am. Coll. Cardiol. 41: 753–760
Mauney M and Kron I (1995) The physiologic basis of warm cardioplegia. Ann. Thorac. Surg. 60: 819–823
Aebert H, Kirchner S and Keyser A (1997) Induction of early immediate genes and programmed cell death following cardioplegic arrest in human hearts. Eur. J. Cardiothorac. Surg. 12: 261–267
Chen Y, Wu S and Huang C (2001) Morphometric identification of luminal narrowing of myocardial capillaries after cardioplegic arrest. Ann. Thorac. Surg. 71: 243–248
Rainio P, Kaukoranta P and Sormunen R (1998) Ultrastructural changes in myocardium during mild hypothermic retrograde blood cardioplegia. Scand. Cardiovasc. J. 32: 353–359
Pelletier LC, Carrier M, Leclerc Y, Cartier R, Wesolowska E and Solymoss BC (1994) Intermittent antegrade warm versus cold blood cardioplegia: a prospective, randomized study. Ann. Thorac. Surg. 58: 41–48
Franke UF, Korsch S, Wittwer T, Albes JM, Wippermann J, Kaluza M, Rahmanian PB and Wahlers T (2003) Intermittent antegrade warm myocardial protection compared to intermittent cold blood cardioplegia in elective coronary surgery – do we have to change? Eur. J. Cardiothorac. Surg. 23: 341–346
Scarabelli TM, Pasini E, Ferrari G, Ferrari M, Stephanou A, Lawrence K, Townsend P, Chen-Scarabelli C, Gitti G, Saravolatz L, Latchman D, Knight R and Gardin J (2004) Warm blood cardioplegic arrest induces mitochondrial-mediated cardiomyocyte apoptosis associated with increased urocortin expression in viable cells. J. Thorac. Cardiovasc. Surg. 128: 364–371
Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST and van Oers MH (1994) Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84: 1415–1420
Tait JF, Cerqueira MD, Dewhurst TA, Fujikawa K, Ritchie JL and Stratton JR (1994) Evaluation of annexin V as a platelet-directed thrombus targeting agent. Thromb. Res. 75: 491–501
Stratton JR, Dewhurst TA, Kasina S, Reno JM, Cerqueira MD, Baskin DG and Tait JF (1995) Selective uptake of radiolabeled annexin V on acute porcine left atrial thrombi. Circulation 92: 3113–3121
Blankenberg FG, Katsikis PD, Tait JF, Davis RE, Naumovski L, Ohtsuki K, Kopiwoda S, Abrams MJ, Darkes M, Robbins RC, Maecker HT and Strauss HW (1998) In vivo detection and imaging of phosphatidylserine expression during programmed cell death. Proc. Natl. Acad. Sci. USA 95: 6349–6354
Dumont EA, Hofstra L, van Heerde WL, van den Eijnde S, Doevendans PA, DeMuinck E, Daemen MA, Smits JF, Frederik P, Wellens HJ, Daemen MJ and Reutelingsperger CP (2000) Cardiomyocyte death induced by myocardial ischemia and reperfusion: measurement with recombinant human annexin-V in a mouse model. Circulation 102: 1564–1568
Dumont EA, Reutelingsperger CP, Smits JF, Daemen MJ, Doevendans PA, Wellens HJ and Hofstra L (2001) Real-time imaging of apoptotic cell-membrane changes at the single-cell level in the beating murine heart. Nat. Med. 7: 1352–1355
Hofstra L, Liem IH, Dumont EA, Boersma HH, van Heerde WL, Doevendans PA, De Muinck E, Wellens HJ, Kemerink GJ, Reutelingsperger CP and Heidendal GA (2000) Visualisation of cell death in vivo in patients with acute myocardial infarction. Lancet 356: 209–212
Kloner RA, Bolli R, Marban E, Reinlib L and Braunwald E (1998) Medical and cellular implications of stunning, hibernation, and preconditioning: an NHLBI workshop. Circulation 97: 1848–1867
Cigarroa CG, deFilippi CR, Brickner ME, Alvarez LG, Wait MA and Grayburn PA (1993) Dobutamine stress echocardiography identifies hibernating myocardium and predicts recovery of left ventricular function after coronary revascularization. Circulation 88: 430–436
Maes A, Flameng W, Nuyts J, Borgers M, Shivalkar B, Ausma J, Bormans G, Schiepers C, De Roo M and Mortelmans L (1994) Histological alterations in chronically hypoperfused myocardium. Correlation with PET findings. Circulation 90: 735–745
Topol EJ, Weiss JL, Guzman PA, Dorsey-Lima S, Blanck TJ, Humphrey LS, Baumgartner WA, Flaherty JT and Reitz BA (1984) Immediate improvement of dysfunctional myocardial segments after coronary revascularization: detection by intraoperative transesophageal echocardiography. J. Am. Coll. Cardiol. 4: 1123–1134
Ausma J, Cleutjens J, Thone F, Flameng W, Ramaekers F and Borgers M (1995) Chronic hibernating myocardium: interstitial changes. Mol. Cell. Biochem. 147: 35–42
Chen C, Ma L, Linfert DR, Lai T, Fallon JT, Gillam LD, Waters DD and Tsongalis GJ (1997) Myocardial cell death and apoptosis in hibernating myocardium. J. Am. Coll. Cardiol. 1;30: 1407–1412
Elsasser A, Schlepper M, Klovekorn WP, Cai WJ, Zimmermann R, Muller KD, Strasser R, Kostin S, Gagel C, Munkel B, Schaper W and Schaper J (1997) Hibernating myocardium: an incomplete adaptation to ischemia. Circulation 96: 2920–2931
Schwarz ER, Schoendube FA, Kostin S, Schmiedtke N, Schulz G, Buell U, Messmer BJ, Morrison J, Hanrath P and vom Dahl J (1998) Prolonged myocardial hibernation exacerbates cardiomyocyte degeneration and impairs recovery of function after revascularization. J. Am. Coll. Cardiol. 31: 1018–1026
Holly TA, Drincic A, Byun Y, Nakamura S, Harris K, Klocke FJ and Cryns VL (1999) Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J. Mol. Cell. Cardiol. 31: 1709–1715
Yaoita H, Ogawa K, Maehara K and Maruyama Y (1998) Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 97: 276–281
Scarabelli TM, Stephanou A, Pasini E, Gitti G, Townsend P, Lawrence K, Chen-Scarabelli C, Saravolatz L, Latchman D, Knight R and Gardin J (2004) Minocycline inhibits caspase activation and reactivation, increases the ratio of XIAP to smac/DIABLO, and reduces the mitochondrial leakage of cytochrome C and smac/DIABLO. J. Am. Coll. Cardiol. 43: 865–874
Chen Z, Chua CC, Ho YS, Hamdy RC and Chua BH (2001) Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 280: H2313–H2320
Cain BS, Meldrum DR, Dinarello CA, Meng X, Banerjee A and Harken AH (1998) Adenosine reduces cardiac TNF-alpha production and human myocardial injury following ischemia–reperfusion. J. Surg. Res. 76: 117–123
Peng CF, Lee P and Deguznman A (2001) Multiple independent mutations in apoptotic signaling pathways markedly decrease infarct size due to myocardial ischemia–reperfusion In Scientific Sessions 2001 Willerson JT (ed) Anaheim, CA: Lippincott
Communal C, Sumandea M, de Tombe P, Narula J, Solaro RJ and Hajjar RJ (2002) Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. USA 99: 6252–6256
Chen-Scarabelli C, Hughes S, Landon G, Rowley P, Allebban Z, Lawson N, Saravolatz L, Gardin J and Scarabelli TM (2004) A case of fatal ephedra intake associated with lipofuscin accumulation, caspase activation and cleavage of myofibrillary proteins. Eur. J. Heart Failure (in press)
Ricci JE, Gottlieb RA and Green DR (2003) Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell Biol. 160: 65–75
Mehta JL and Li DY (1999) Inflammation in ischemic heart disease: response to tissue injury or a pathetic villain? Cardiovasc. Res. 2: 291–299
Maroko PR, Carpenter CB, Chiariello M, Fishbein MC, Radvany P, Knostman JD and Hale SL (1978) Reduction by cobra venom factor of myocardial necrosis after coronary artery occlusion. J. Clin. Invest. 3: 661–670
Shappell SB, Taylor AA, Hughes H, Mitchell JR, Anderson DC and Smith CW (1990) Comparison of antioxidant and nonantioxidant lipoxygenase inhibitors on neutrophil function. Implications for pathogenesis of myocardial reperfusion injury. J. Pharmacol. Exp. Ther. 2: 531–538
Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA and Lucchesi BR (1983) Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation 5: 1016–1023
Mullane KM, Read N, Salmon JA and Moncada S (1984) Role of leukocytes in acute myocardial infarction in anesthetized dogs: relationship to myocardial salvage by anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2: 510–522
Engler RL, Dahlgren MD, Morris DD, Peterson MA and Schmidt-Schoenbein GW (1986) Role of leukocytes in response to acute myocardial ischemia and reflow in dogs. Am. J. Physiol. 251 (Part 2): H314–H323
Jolly SR, Kane WJ, Bailie MB, Abrams GD and Lucchesi BR (1984) Canine myocardial reperfusion injury. Its reduction by the combined administration of superoxide dismutase and catalase. Circ. Res. 3: 277–285
Giugliano GR, Giugliano RP, Gibson CM and Kuntz RE (2003) Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am. J. Cardiol. 91: 1055–1059
Ko D, Wang Y, Berger AK, Radford MJ and Krumholz HM (2002) Nonsteroidal antiinflammatory drugs after acute myocardial infarction. Am. Heart J. 143: 475–481
Weisman HF, Bush DE, Mannisi JA and Weisfeldt ML (1988) Healy B. Cellular mechanism of myocardial infarct expansion. Circulation 78: 186–201
Jugdutt BI and Michorowski BL (1987) Role of infarct expansion in rupture of ventricular septum after acute myocardial infarction: a two dimensional echocardiographic study. Clin. Cardiol. 10: 641–652
Weisman HF and Healy B (1987) Myocardial infarct expansion, infarct extension, and reinfarction: pathophysiologic concepts. Prog. Cardiovasc. Dis. 30: 73–110
Schuster EH and Bulkley BH (1979) Expansion of transmural myocardial infarction: a pathophysiologic feature in cardiac rupture. Circulation 60: 1532
Gottlieb RA, Gruol DL, Zhu JY and Engler RL (1996) Preconditioning rabbit cardiomyocytes: role of pH, vacuolar proton ATPase, and apoptosis. J. Clin. Invest. 97: 2391–2398
Scarabelli TM, Pasini E, Stephanou A, Comini L, Curello S, Raddino R, Ferrari R, Knight R and Latchman DS (2002) Urocortin promotes hemodynamic and bioenergetic recovery and improves cell survival in the isolated rat heart exposed to ischemia/reperfusion. J. Am. Coll. Cardiol. 40: 155–161
Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E and Mochly-Rosen D (2003) Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation 108: 2304–2307
Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth Jr P, Yeager M and Gottlieb RA (2004) Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc. Natl. Acad. Sci. USA 101: 1321–1326
Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, Burstein D, Doukas J, Soll R, Losordo D and Cheresh D (2004) Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J. Clin. Invest. 113: 885–894
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by RA Knight
Rights and permissions
About this article
Cite this article
Scarabelli, T., Gottlieb, R. Functional and clinical repercussions of myocyte apoptosis in the multifaceted damage by ischemia/reperfusion injury: old and new concepts after 10 years of contributions. Cell Death Differ 11 (Suppl 2), S144–S152 (2004). https://doi.org/10.1038/sj.cdd.4401544
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401544
Keywords
This article is cited by
-
Cardioprotective effect of ghrelin against myocardial infarction-induced left ventricular injury via inhibition of SOCS3 and activation of JAK2/STAT3 signaling
Basic Research in Cardiology (2018)
-
Nicotine plus a high-fat diet triggers cardiomyocyte apoptosis
Cell and Tissue Research (2017)
-
Mitochondrial sirtuins in the heart
Heart Failure Reviews (2016)
-
Regenerating cardiac cells: insights from the bench and the clinic
Cell and Tissue Research (2012)
-
Minocycline suppresses oxidative stress and attenuates fetal cardiac myocyte apoptosis triggered by in utero cocaine exposure
Apoptosis (2011)
