
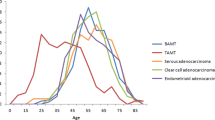
Abstract
Goblet cell carcinoid is a relatively rare neuroendocrine tumor of the vermiform appendix with poorly understood molecular pathogenesis. We studied the clinicopathologic features and genetic alterations, including allelic loss of chromosomes 11q, 16q, and 18q; sequencing of the K-ras, β-catenin, and DPC4 (SMAD4) genes; and p53 overexpression and loss of DPC4 by immunohistochemistry; in 16 goblet cell carcinoids. We compared the allelic loss in goblet cell carcinoids to those in 18 gastrointestinal carcinoid tumors. For goblet cell carcinoids, appendiceal perforation was the most common (70%, 7/10) clinical presentation. The mean tumor size was 2.0 ± 1.5 cm (range, 0.4 to 4.5 cm). The tumor invaded to appendiceal serosa in 50% (8/16) of patients, and two patients had metastasis in lymph nodes or adjoining viscera. With mean follow-up of 24 ± 14 months (median, 23 mo), 1 of 10 patients had died of disease, and 2 others had tumor recurrence. All four patients with metastases, recurrences, and/or death from disease had serosal involvement at presentation (P = .02). Loss of heterozygosity of chromosome 11q was present in 25% of goblet cell carcinoids, 14% of ileal carcinoid tumors, and 9% of nonileal carcinoid tumors; of chromosome 16q in 38%, 29%, and 0 (P = .02); and of chromosome 18q in 56%, 86%, and 9% (P = .002), respectively. No mutations of K-ras, β-catenin, or DPC4 genes; p53 overexpression; or loss of staining for DPC4 was present in any tumors. These findings suggest that allelic loss of chromosomes 11q, 16q, and 18q in goblet cell carcinoids and ileal carcinoids may have an important role in the pathogenesis of these tumors.
Similar content being viewed by others
Main
Goblet cell carcinoid (International Classification of Diseases for Oncology, ICD-O 8243/3) is a relatively rare neuroendocrine tumor of the vermiform appendix, comprising approximately 6% of appendiceal carcinoids (1, 2). First described in 1974 (3), goblet cell carcinoid is currently considered an amphicrine tumor with histopathologic and ultrastructural features that overlap between typical carcinoid and adenocarcinoma (2). Although goblet cell carcinoid was previously considered a variant of carcinoid tumor, current evidence suggests that goblet cell carcinoid is a distinct tumor with a histogenesis different from that of the typical carcinoid, reportedly arising from the multipotential epithelial crypt stem cells (4, 5). In contrast, the typical carcinoid tumor of the appendix is thought to originate in the subepithelial neuroendocrine cells of neural crest derivation (6, 7). Furthermore, the clinical behavior of appendiceal goblet cell carcinoid is considered intermediate between that of typical carcinoid tumors and that of appendiceal adenocarcinomas, with an overall 5-year survival rate of 73–84% (8, 9). All these clinical and histopathologic data suggest that goblet cell carcinoid is a distinctive clinicopathologic entity.
Because of its rarity, little is known about the molecular pathogenesis of goblet cell carcinoid. In contrast, rapid advances have been made in characterization of molecular events underlying the tumorigenesis of related tumors, such as typical carcinoids and adenocarcinomas of the intestinal tract. Whereas mutations of K-ras proto-oncogene are frequent in adenocarcinomas of the colorectum (10, 11), appendix (12, 13, 14), and duodenum (15, 16), they are absent in typical carcinoid tumors of appendix and small bowel and in appendiceal goblet cell carcinoids (15, 17, 18). More controversial are the results reported for alterations in goblet cell carcinoids of the p53 gene, a tumor suppressor gene frequently altered in colorectal neoplasia. One study reported the presence of p53 gene point mutations without immunohistochemical detection of p53 in both typical carcinoid tumors and goblet cell carcinoids of the appendix (18). In contrast, another study failed to demonstrate such alterations (17). Deregulation of β-catenin signaling is an important event in colorectal carcinoma (19, 20, 21). Overexpression of β-catenin leads to its translocation to cell nucleus together with Tcf/Lef-1 and formation of a bipartite transcriptional factor that activates transcription of many target genes, including c-myc and cyclin D1 (20). Allelic loss of chromosomes 18q, 16q, and 11q is present in typical midgut carcinoids (22), and allelic loss of chromosome 18q is frequent in colorectal carcinomas (23). DPC4 (Smad4) gene mutations are present in about 50% of pancreatic (24) and in about 20% of colorectal carcinomas (25). However, the role of β-catenin and DPC4 in the pathogenesis of goblet cell carcinoid is unknown.
Because these genetic alterations have not been assessed in appendiceal goblet cell carcinoid, we studied cases for mutations of K-ras, β-catenin, and DPC4 genes; p53 overexpression, and loss of DPC4 staining by using immunohistochemistry; and allelic loss of chromosomes 11q, 16q, and 18q as well as clinicopathological associations.
MATERIALS AND METHODS
Case Material
Sixteen appendiceal goblet cell carcinoids (ICD-O 8243/3) were identified from the surgical pathology files of the University of Texas M.D. Anderson Cancer Center from patients undergoing resection between 1991 and 2002 and available formalin-fixed, paraffin-embedded tissue blocks (1). The patient records and histopathological findings were reviewed. The diagnosis of goblet cell carcinoid was confirmed in all cases using the World Health Organization classification of appendiceal tumors (1). Mixed adenocarcinoma-carcinoid tumors were excluded from the study. Goblet cell carcinoids were compared with 18 gastrointestinal carcinoid tumors, including 15 tumors from a previously reported study (26) and 3 appendiceal carcinoid tumors. The study was approved by the Surveillance Committee (institutional review board) of the University of Texas M.D. Anderson Cancer Center.
DNA Preparation
Genomic DNA was extracted from tumor and control nonneoplastic tissues after microdissection from 5-μm sections of formalin-fixed, paraffin-embedded tissue as described previously (27).
LOH of Chromosomes 11q, 16q, and 18q
LOH of chromosomes 11q, 16q, and 18q was determined by use of dinucleotide microsatellite markers with PCR amplification using fluorescent dye–labeled primers (Invitrogen, Carlsbad, CA). Chromosome 11q was assessed by microsatellite markers D11S4191, D11S937, D11S4127, D11S912, and D11S968; chromosome 16q by D16S3136, D16S514, D16S3066, D16S505, and D16S520; and chromosome 18q by D18S474, D18S69, D18S64, D18S1147, D18S55, D18S61, D18S58, and D18S70, respectively. The 5′oligonucleotide was end-labeled with 6-FAM fluorescent dye. PCR was performed in 15-μL reaction volumes containing 20 ng of DNA, 9 μL of ABI Prism True Allele PCR Premix (Applied Biosystems, Foster City, CA), and 5 pmol of each primer. The cycling conditions were denaturation at 95° C for 12 minutes, 10 cycles (94° C for 15 s, 55° C for 15 s, 72° C for 30 s), 32 cycles (89° C for 15 s, 55° C for 15 s, 72° C for 30 s), and extension at 72° C for 10 minutes. A 0.25-μL aliquot of each fluorescent-labeled PCR product was combined with 12 μL of formamide and 0.5 μL of GENESCAN 400HD [ROX] size standard (Applied Biosystems). The samples were then subjected to capillary electrophoresis on an ABI 3700 DNA Analyzer by using GENESCAN Analysis software (Applied Biosystems). Loss of a marker was considered to be present when the assay showed absence or decrease in intensity by >50% of one of two alleles from a tumor sample as compared with the paired control nonneoplastic tissue. LOH of chromosomal arms was defined by allelic loss of one or more polymorphic (informative) microsatellite markers present on that chromosomal arm.
DNA Sequencing of Exon 1 of K-Ras, Exon 3 of β-Catenin, and Exons 8–11 of DPC4 Genes
Exon 1 of the K-ras proto-oncogene, exon 3 of the β-catenin gene, and exons 8–11 of the DPC4 gene were amplified by PCR and sequenced using the primers and PCR conditions as described previously (28, 29, 30). DNA sequencing was performed on the PCR product and amplification primers for each exon with a commercial DNA sequencing kit according to the manufacturer’s instructions (Applied Biosystems).
Immunohistochemistry for p53 and DPC4
Expression of p53 gene was assessed by using mouse monoclonal antibody D07 (1:250 dilution) and standard techniques including antigen retrieval as described previously (31). Overexpression for p53 gene product was considered when >50% of the tumor cells demonstrated nuclear staining by immunohistochemistry.
DPC4 protein staining was assessed by using mouse monoclonal antibody to DPC4 (clone B8, 1:250 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) and standard techniques including antigen retrieval as described previously (32). Positive staining for DPC4 protein was scored as diffusely positive if >50% of tumor cells were staining, focally positive if <50% of cells were positive, or negative if no tumor cells were positive. Normal epithelium, fibroblasts, and lymphocytes served as positive internal controls in each of the sections.
Statistical Analysis
The differences in frequencies among clinicopathological factors and genetic alterations, and between genetic alterations of goblet cell carcinoids and carcinoid tumors were analyzed by Fisher’s exact test. All statistical analyses were performed using the SPSS statistical program (SPSS Inc., Chicago, IL).
RESULTS
Clinicopathologic Features
The clinicopathological features of the 16 patients with goblet cell carcinoid are summarized in Table 1. There were 8 male and 8 female patients, ranging from 23 to 74 years in age (mean, 47 ± 16 y). Perforated appendix was the most common clinical presentation and was present in 70% (7 of 10) patients. In two patients the tumor was detected incidentally.
Ten patients had appendectomy, including 1 patient who had total abdominal hysterectomy with bilateral salpingo-oophorectomy, 1 patient had appendectomy and regional lymph node dissection, 2 patients had appendectomy followed by right ileocolectomy, and 3 patients had right ileocolectomy. The mean size of tumor was 2.0 ± 1.5 cm (range, 0.4 to 4.5 cm). Histologically, all tumors were composed of scattered nests and tubules of small-to-intermediate size cells with monomorphic nuclei and scant eosinophilic cytoplasm, admixed with goblet cells (Fig. 1). In all cases, an infiltrative single-file growth pattern of bland goblet cells comprised 25% or less of the dominant (nested) tumor growth pattern. At least focal immunohistochemical staining for chromogranin A and synaptophysin was detected in all tumors. The tumor invaded the serosa of the appendix in 50% (8/16) of patients, the subserosal connective tissue of appendix or mesoappendix in 31% (5 of 16) patients, or the muscularis propria of appendix in 13% (2 of 16) patients. Two patients had regional lymph nodes metastases (Patients 1 and 4), and one (Patient 1) also had omental metastases.

Goblet cell carcinoid of the appendix. (Original magnification 200 ×).
A mean clinical follow-up of 24 ± 14 months (median, 23 mo) was available for 10 patients. All but 2 patients were alive. One patient with regional tumor spread and lymph node metastasis (Patient 1) died of goblet cell carcinoid 24 months after diagnosis. The second patient (Patient 15) died of an unrelated malignancy 36 months after diagnosis. Two other patients had recurrences: in the small bowel, sigmoid colon, peritoneum, and abdominal wall (Patient 2) at 20 months after diagnosis or in omentum (Patient 3) at 32 months after diagnosis. All four patients with lymph node metastases, recurrence and/or death due to goblet cell carcinoid had serosal involvement of the appendix at presentation (4/8 versus 0/8, P = .02). Two patients had appendectomy followed by right ileocolectomy, and minimal residual disease (small subserosal tumor deposit in the cecum adjacent to appendectomy site) was found in one of these (Patient 2). This patient received postoperative chemotherapy with 5-fluorouracil, leucovorin, and CPT-11 and was alive with disease 34 months after diagnosis.
The clinicopathological features of carcinoid tumors are summarized in Table 2.
Allelic Loss in Goblet Cell Carcinoids
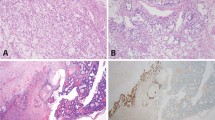
LOH of chromosome 11q was present in 27% (4/15) of goblet cell carcinoids (examples in Fig. 2 and summarized in Table 3). The markers D11S937 and D11S968 showed loss in 8% and 50% of informative loci, respectively, but adjoining centromeric or telomeric markers D11S4191, D11S4127, and D11S912 did not show any loss. LOH of chromosome 16q was present in 37% (6/16) of goblet cell carcinoids. The telomeric markers D16S3066, D16S505, and D16S520 showed loss in 37%, 20%, and 9% of informative loci, respectively, but centromeric markers D16S3136 and D16S514 did not show any loss. LOH of chromosome 18q was present in 56% (9/16) of goblet cell carcinoids. The telomeric markers D18S55, D18S61, D18S58, and D18S70 showed loss in 66%, 30%, 50%, and 25% of informative loci, respectively. Five goblet cell carcinoids showed loss of centromeric markers D18S474, D18S69, and D18S64, and two of these tumors had no loss of telomeric markers and one had retention of an intervening marker. Fraction allelic loss (LOH of polymorphic markers/number of polymorphic markers for each tumor) ranged from 0 to 62% (mean, 27.8%). No correlation was found between LOH status and the clinicopathologic features of goblet cell carcinoids.

Chromosome 11, 16q and 18q loss by PCR amplification using fluorescent primers in goblet cell carcinoids and gastrointestinal carcinoid tumors. The markers are indicated at the top. PCR was performed using nonneoplastic DNA (N) and tumor DNA (T). Examples of allelic loss in goblet cell carcinoids (top panels) and gastrointestinal carcinoid tumors (bottom panels) are shown in representative tumors for each marker.
Comparison of Allelic Loss in Goblet Cell Carcinoids with Carcinoid Tumors
LOH of chromosomes 11q, 16q, and 18q was present in 11% (2/18), 11% (2/18), and 39% (7/18) of carcinoid tumors (examples in Fig. 2 and summarized in Table 4). This was predominantly due to frequent LOH in ileal carcinoid tumors compared with nonileal carcinoid tumors (Table 5): loss of chromosomes 11q was present in 14% of ileal carcinoid versus 9% of nonileal carcinoids, loss of chromosome 16q in 29% versus 0% (P = .04), and loss of chromosome 18q in 86% versus 9% (P = .0007). Fraction allelic loss ranged from 0 to 73% (mean, 41.2%) in ileal carcinoid tumors and ranged from 0 to 8% (mean, 1.2%) in carcinoid tumors of other sites.
Allelic loss of chromosomes 11q, 16q, and 18q was as frequent in goblet cell carcinoids as in ileal carcinoid tumors but was less frequent in nonileal carcinoid tumors (Table 5): LOH of chromosome 11q was present in 25% of goblet cell carcinoids, 14% of ileal carcinoid tumors, and 9% of nonileal carcinoid tumors; of chromosome 16q in 38%, 29%, and 0 (P = .02); and of chromosome 18q in 56%, 86%, and 9% (P = .002), respectively.
Other Genetic Alterations of Goblet Cell Carcinoids
No K-ras or β-catenin mutations in 16 tumors, DPC4 mutations in 15 tumors, p53 overexpression in 12 tumors, or loss of staining for DPC4 in 14 tumors were identified.
DISCUSSION
In our study, we found infrequent allelic loss of chromosome 11q but frequent allelic loss of chromosomes 16q and 18q in goblet cell carcinoids and carcinoid tumors. The long arm of chromosome 11 is a gene-rich region (33) but contains only a few known or putative tumor suppressor genes mapped to 11q13 (MEN 1, SDHD and PGL2 genes) and 11q22-q23 (PGL1 and PPP2R1B genes). Allelic loss of more distal chromosomal regions located at 11q25 were identified in breast and ovarian cancer (33). Similar to our study, in a recent report, allelic loss of 11q25 was identified in 40% of classical ileal carcinoids and was not found in any of the seven classical appendiceal carcinoids (34). Another study reported a high frequency of allelic loss at 11q13 locus (62%) in classical midgut carcinoids, but a low frequency of SDHD gene mutations, suggesting that other tumor suppressor genes may be targets for allelic loss at this region (35). In our study, allelic loss of chromosome 11q in goblet cell carcinoids was present at locus 11q25, and only one GCC and one ileal carcinoid tumor had loss of 11q13 locus, suggesting that MEN 1 or SDHD genes are not involved in the tumorigenesis of appendiceal goblet cell carcinoids. These findings suggest that an unknown tumor suppressor gene located on 11qter may have a role in tumorigenesis of some of the appendiceal goblet cell carcinoid and in ileal carcinoid tumors.
More than a third of goblet cell carcinoids and ileal carcinoid tumors in our study demonstrated LOH of 16q with loss of loci 16q21-q24.2. This chromosomal region harbors two putative tumor suppressor genes: CTCF gene (CCCTC-binding factor), a transcriptional repressor of c-myc, and the E-cadherin/CDH1 gene involved in cell adhesion. Homozygous loss of function of CTCF gene by tumor-specific rearrangements has been identified in breast carcinomas (36), whereas mutations in E-cadherin have been reported in invasive lobular carcinoma of breast (37). Interestingly, Kytola et al. (22) reported allelic loss of 16q21-qter in metastatic midgut carcinoids but not in primary carcinoid tumors, suggesting that putative tumor suppressor genes mapped at this locus may be involved in tumor progression.
In our study we identified frequent allelic loss of chromosome 18q in goblet cell carcinoids and ileal carcinoid tumors, and no loss of 18p in goblet cell carcinoids. The high frequency of allelic loss of 18q in our cases indicates that the genetic alterations (allelic loss or mutational inactivation) of tumor suppressor genes mapped to the telomeric 18q play an important role in the carcinogenesis of appendiceal goblet cell carcinoids and ileal carcinoid tumors. The most frequent chromosomal loss was on the long arm of chromosome 18 at the locus 18q21–23 in goblet cell carcinoids that is frequently deleted in different types of tumors including pancreatic, colorectal, and breast carcinomas (for review, see (38)). In contrast, the ileal carcinoid tumors had allelic loss of all the markers on chromosome 18q or loss of two or three loci suggesting involvement of more than one gene on chromosome 18q. Among the tumor suppressor genes harbored on 18q, the most extensively studied are the DCC gene localized at 18q21.2 and the DPC4 (SMAD4) gene localized at 18q21.1. Although inactivation of DCC is identified not only in colorectal carcinoma (39), but also in pancreatic, gastric, mammary, prostatic carcinomas (40, 41, 42, 43), DPC4 gene inactivation occurs in almost half of the pancreatic carcinomas (24) but is uncommon in other malignancies (44, 45). We ruled out involvement of DPC4 gene by demonstrating no DPC4 mutations or loss of staining of DPC4 protein by immunohistochemistry. However, the role of DCC gene in GCCs remains unclear. Other putative tumor suppressor genes localized immediately telomeric to DCC and DPC4 at microsatellite locus 18q21.3 (D18S55) are serpins (serine proteinase inhibitors), some of which (maspin) reportedly can suppress tumor growth and spread in mammary carcinoma (46). In our study, allelic loss at loci 18q21–23 that are telomeric to DCC and DPC4 genes suggests loss of a region that harbors some of the cadherin genes that are considered tumor suppressor genes because of their role in cell adhesion (47). Interestingly, a similar frequency of allelic loss of 18q was reported for classical midgut carcinoids (22, 48) and for sporadic adenocarcinomas of the small intestine (49) but not in two studies of lung carcinoids (50, 51). However, ileal carcinoids lack DPC4 mutations as compared with small intestinal carcinomas (48, 49).
In our study, there were no K-ras, β-catenin, or DPC4 mutations; p53 overexpression; or loss of staining for DPC4 in goblet cell carcinoids. K-ras oncogene was shown to have a major role in carcinogenesis and/or tumor progression in a wide variety of human neoplasms including colorectal carcinomas, pancreatic adenocarcinomas, cholangiocarcinomas, and lung adenocacinomas (52). Recently, Kabbani et al. (12) reported frequent K-ras mutations in appendiceal adenocarcinomas. In contrast, we and other investigators (18) did not find K-ras mutations in goblet cell carcinoids. Furthermore, K-ras mutations are not present in foregut or midgut carcinoid tumors or pancreatic endocrine tumors (15, 17, 18, 53).
A recent study reported abnormal nuclear accumulation of β-catenin in 36% digestive tract carcinoids and 27% of islet cell tumors (54), but no genetic alteration of β-catenin (CTNNB1) gene (26, 54). Li et al. (55) found that the majority of the goblet cell carcinoids of the appendix studied demonstrated strong membranous staining for β-catenin, similar to normal appendiceal mucosa, and no nuclear or cytoplasmic staining. In our study, none of the appendiceal goblet cell carcinoid demonstrated mutations of β-catenin gene.
p53 overexpression was not identified in goblet cell carcinoids in our study, a finding that is in agreement with previous reports of infrequent p53 gene mutations in goblet cell carcinoid and adenocarcinomas of appendix (2, 12, 17) In contrast, Ramnani et al. (18) reported p53 mutations in 25% and 44% of appendiceal goblet cell and classical carcinoids, respectively.
Overall, these findings suggest that alterations of oncogenes and tumor suppressor genes, frequently altered in adenocarcinomas of gastrointestinal tract, do not play a role in the pathogenesis of appendiceal goblet cell carcinoid.
Goblet cell carcinoid is considered a malignancy that is more aggressive than carcinoid tumor of the appendix (2, 8, 9, 56, 57, 58), with 5-year survival rate ranging from 73 to 84% (8, 9). In our study, serosal involvement by the tumor at presentation was associated with adverse outcome, similar to the case in previously reported studies (8, 18, 56), suggesting that tumor stage is the most important prognostic factor in appendiceal goblet cell carcinoid. Another possible unfavorable prognostic factor reported by Burke and co-workers (59) is the percentage of carcinomatous growth defined as fused or cribriform glands, single-file structures, diffusely infiltrating signet-ring cells, or solid sheets of cells. According to this study, all 25 patients with appendiceal goblet cell carcinoids with no or <25% carcinomatous growth were alive an average of 19 months after diagnosis, regardless of the treatment modality. However, this finding may not be conclusive because goblet cell carcinoid of the appendix may recur and metastasize >15 years after diagnosis (2, 58).
In conclusion, our study provides evidence that goblet cell carcinoids of the appendix are neuroendocrine tumors with genetic alterations similar to those of ileal carcinoid tumors and different from those of appendiceal adenocarcinomas. The loss of chromosomes 11q, 16q, and 18q identified in the majority of these tumors suggests that putative or yet unknown tumor suppressor genes may have a major role in the pathogenesis of goblet cell carcinoids and ileal carcinoids.
References
Capella C, Solcia E, Sobin LH, Arnold R . Endocrine tumor of the appendix. In: Hamilton SR, Aaltonen LA, editors. World Health Organization classification of tumors: pathology and genetics of tumors of the digestive system. Lyon, France: IARC Press; 2000. p. 99–101.
Kanthan R, Saxena A, Kanthan SC . Goblet cell carcinoids of the appendix: immunophenotype and ultrastructural study. Arch Pathol Lab Med 2001; 125: 386–390.
Subbuswamy SG, Gibbs NM, Ross CF, Morson BC . Goblet cell carcinoid of the appendix. Cancer 1974; 34: 338–344.
Goddard MJ, Lonsdale RN . The histogenesis of appendiceal carcinoid tumors. Histopathology 1992; 20: 345–349.
Isaacson P . Crypt cell carcinoma of the appendix (so-called adenocarcinoid tumor). Am J Surg Pathol 1984; 5: 213–224.
Lundqvist M, Wilander E . Subepithelial neuroendocrine cells and carcinoid tumors of the human small intestine and appendix. A comparative immunohistochemical study with regard to serotonin, neuron specific enolase and S-100 protein specificity. J Pathol 1986; 148: 141–147.
Rhode J, Dhillon AP, Papadaki L, Griffiths D . Neurosecretory cells of the lamina propria of the appendix and their possible relationship to carcinoids. Histopathology 1982; 6: 69–79.
Warkel RL, Cooper PH, Helwig EB . Adenocarcinoid, a mucin-producing carcinoid tumor of the appendix: a study of 39 cases. Cancer 1978; 42: 2781–2793.
Edmonds P, Merino MJ, LiVolsi VA, Duray PH . Adenocarcinoid (mucinous carcinoid) of the appendix. Gastroenterology 1984; 86: 302–309.
Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der EB AJ, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987; 327: 293–297.
Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988; 319: 525–532.
Kabbani W, Houlihan PS, Luthra R, Hamilton SR, Rashid A . Mucinous and nonmucinous appendiceal adenocarcinomas: different clinicopathological features but similar genetic alterations. Mod Pathol 2002; 15: 599–605.
Cuatrecasas M, Matias-Guiu X, Prat J . Synchronous mucinous tumors of the appendix and the ovary associated with pseudomyxoma peritonei: a clinicopathologic study of six cases with comparative c-Ki-ras mutations. Am J Surg Pathol 1996; 20: 739–746.
Szyck C, Staebler A, Connolly DC, Wu R, Cho KR, Ronnett BM . Molecular genetic evidence supporting the clonality and appendiceal origin of pseudomyxoma peritonei in women. Am J Pathol 1999; 154: 1849–1855.
Younes N, Fulton N, Tanaka R, Wayne J, Strauss FH II, Kaplan EL . The presence of K-12 ras mutations in duodenal adenocarcinomas and the absence of ras mutations in other small bowel adenocarcinomas and carcinoid tumors. Cancer 1997; 79: 1804–1808.
Sutter T, Arber N, Moss SF, Findling RI, Neugut AI, Weinstein IB, et al. Frequent K-ras mutations in small bowel adenocarcinomas. Dig Dis Sci 1996; 41: 115–118.
Paraskevakou H, Saetta A, Skandalis K, Tseleni S, Athanassiadis A, Davaris PS . Morphological-histochemical study of intestinal carcinoids and K-ras mutation analysis in appendiceal carcinoids. Pathol Oncol Res 1999; 5: 205–210.
Ramnani DM, Wistuba II, Behrens C, Gazdar AF, Sobin LH, Albores-Saavedra J . K-ras and p53 mutations in the pathogenesis of classical and goblet cell carcinoids of the appendix. Cancer 1999; 86: 14–21.
Hiscox S, Jiang WG . Expression of E-cadherin, alpha, beta and gamma-catenin in human colorectal cancer. Anticancer Res 1997; 17: 1349–1354.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997; 275: 1787–1790.
Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, et al. Constitutive transcriptional activation by beta-catenin-Tcf complex in APC-/-colon carcinoma. Science 1997; 275: 1752–1753.
Kytölä S, Höög A, Nord B, Cedermark B, Frisk T, Larsson C, et al. Comparative genomic hybridization identifies loss of 18q22-qter as an early and specific event in tumorigenesis of midgut carcinoids. Am J Pathol 2001; 158: 1803–1808.
Watanabe T, Wu TT, Catalano PJ, Ueki T, Satiano R, Haller DG, et al. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med 2001; 344: 1196–1206.
Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996; 271: 350–353.
Ohtaki N, Yamaguchi A, Goi T, Fukaya T, Takeuchi K, Katayama K, et al. Somatic alterations of the DPC4 and Madr2 genes in colorectal cancers and relationship to metastasis. In J Oncol 2001; 18: 265–270.
Chan AO, Kim SG, Bedeir A, Issa JP, Hamilton SR, Rashid A . CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene 2003; 22: 924–934.
Moskaluk CA, Kern SE . Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997; 150: 1547–1552.
Chan AO, Broaddus RR, Houlihan PS, Issa JP, Hamilton SR, Rashid A . CpG island methylation in aberrant crypt foci of the colorectum. Am J Pathol 2002; 160: 1823–1830.
Rashid A, Gao YT, Bhakta S, Shen MC, Wang BS, Deng J, et al. Beta-catenin mutations in billiary tract cancers: a population-based study in China. Cancer Res 2001; 61: 3406–3409.
Hahn SA, Bartsch D, Schroers A, Galehdari H, Becker M, Ramaswamy A, et al. Mutations of DPC4/Smad4 gene in biliary tract carcinoma. Cancer Res 1998; 58: 1124–1126.
Baas IO, Mulder JW, Offerhaus GJ, Vogelstein B, Hamilton SR . An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol 1994; 172: 5–12.
Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, et al. Immunohistochemical labeling for Dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol 2000; 150: 37–43.
Koreth J, Bakkenist CJ, McGee JO . Chromosomes, 11q and cancer: a review. J Pathol 1999; 187: 28–38.
D’adda T, Pizzi S, Azzoni C, Bottarelli L, Crafa P, Pasquali C, et al. Different patterns of 11q allelic losses in digestive endocrine tumors. Hum Pathol 2002; 33: 322–329.
Kytölä S, Nord B, Elder EE, Carling T, Kjellman M, Cedermark B, et al. Alterations of SDHD gene locus in midgut carcinoids, Merkel cell carcinomas, pheochromocytomas, and abdominal paragangliomas. Genes Chromosomes Cancer 2002; 34: 325–332.
Filippova GN, Lindblom A, Meincke LJ, Klenova EM, Neiman PE, Collins SJ, et al. A widely expressed transcription factor with multiple DNA sequence specificity, CTCF, is localized at chromosome segment 16q22.1 within one of the smallest regions of overlap for common deletions in breast and prostate cancers. Genes Chromosomes Cancer 1998; 22: 26–36.
Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, et al. E-cadherin is a tumor/invasion suppressor gene mutated in human lobular breast cancers. EMBO J 1995; 14: 6107–6115.
Knuutila S, Aalto Y, Autio K, Bjorkqvist AM, El-Rifai W, Hemmer S, et al. DNA copy number losses in human neoplasms. Am J Pathol 1999; 155: 683–694.
Fearon ER, Cho KR, Nigro JM, Kern SE, Simons JW, Ruppert JM, et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 1990; 247: 49–56.
Hohne MW, Halatsch ME, Kahl GF, Weinel RJ . Frequent loss of expression of the potential tumor suppressor gene DCC in ductal pancreatic adenocarcinoma. Cancer Res 1992; 52: 2616–2619.
Uchino S, Tsuda H, Noguchi M, Yokota J, Terada M, Saito T, et al. Frequent loss of heterozygosity at the DCC locus in gastric cancer. Cancer Res 1992; 52: 3099–3102.
Devilee P, van Vliet M, Kuipers-Dijkshorn N, Pearson PL, Cornelisse CJ . Somatic genetic changes on chromosome 18 in breast carcinomas: is DCC gene involved? Oncogene 1991; 6: 311–315.
Gao X, Honn KV, Grignon D, Sakr W, Chen YQ . Frequent loss of expression and loss of heterozygosity of the putative tumor suppressor gene DCC in prostatic carcinomas. Cancer Res 1993; 53: 2723–2727.
Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, et al. DPC4 gene in various tumor types. Cancer Res 1996; 56: 2527–2530.
Powell SM, Harper JC, Hamilton SR, Robinson CR, Cummings OW . Inactivation of Smad4 in gastric carcinomas. Cancer Res 1997; 57: 4221–4224.
Zou Z, Anisowicz A, Hendricks MJ, Thor A, Neveu M, Sheng S . Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science 1994; 263: 526–529.
Kremmidiotis G, Baker E, Crawford J, Eyre HJ, Nahmias J, Callen DF . Localization of human cadherin genes to chromosome regions exhibiting cancer-related loss of heterozygosity. Genomics 1998; 49: 467–471.
Lollgen RM, Hessman O, Szabo E, Westin G, Akerstrom G . Chromosome 18 deletions are common events in classical midgut carcinoid tumors. Int J Cancer 2001; 92: 812–815.
Bläker H, von Herbay A, Penzel R, Groβ S, Otto HF . Genetics of adenocarcinomas of the small intestine: frequent deletions at chromosome 18q and mutations of the SMAD4 gene. Oncogene 2002; 21: 158–164.
Walch AK, Zitzelsberger HF, Aubele MM, Mattis AE, Bauchinger M, Candidus S, et al. Typical and atypical carcinoid tumors of the lung are characterized by 11q deletions as detected by comparative genomic hybridization. Am J Pathol 1998; 153: 1089–1098.
Zhao J, de Krijger RR, Meier D, Speel EJ, Saremaslani P, Muletta-Feurer S, et al. Genomic alterations in well-differentiated gastrointestinal and bronchial neuroendocrine tumors (carcinoids): marked differences indicating diversity in molecular pathogenesis. Am J Pathol 2000; 157: 1431–1438.
Rodenhuis S . Ras and human tumors. Semin Cancer Biol 1992; 3: 241–247.
Onuki N, Wistuba II, Travis WD, Virmani AK, Yashima K, Brambilla E, et al. Genetic changes in the spectrum of neuroendocrine lung tumors. Cancer 1999; 85: 600–607.
Semba S, Kusumi R, Moriya T, Sasano H . Nuclear accumulation of β-catenin in human endocrine tumors: association with Ki-67 (mib-1) proliferative activity. Endocr Pathol 2000; 11: 243–250.
Li CC, Hirokawa M, Qian ZR, Xu B, Sano T . Expression of E-cadherin, β-catenin, and Ki-67 in goblet cell carcinoids of the appendix: an immunohistochemical study with clinical correlation. Endocr Pathol 2002; 13: 47–58.
Anderson NH, Somerville JE, Johnston CF, Hayes DM, Buchanan KD, Sloan JM . Appendiceal goblet cell carcinoids: a clinicopathologic and immunohistochemical study. Histopathology 1991; 18: 61–65.
Berardi RS, Lee SS, Chen HP . Goblet cell carcinoids of the appendix. Surg Gynecol Obstet 1988; 167: 81–86.
Park K, Blessing K, Kerr K, Chetty U, Gilmour H . Goblet cell carcinoid of the appendix. Gut 1990; 31: 322–324.
Burke A, Sobin L, Federspiel B, Shekitka K, Helwig E . Goblet cell carcinoids and related tumors of the vermiform appendix. Am J Clin Pathol 1990; 94: 27–35.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stancu, M., Wu, TT., Wallace, C. et al. Genetic Alterations in Goblet Cell Carcinoids of the Vermiform Appendix and Comparison with Gastrointestinal Carcinoid Tumors. Mod Pathol 16, 1189–1198 (2003). https://doi.org/10.1097/01.MP.0000097362.10330.B1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000097362.10330.B1
Keywords
This article is cited by
-
Mutational landscape of goblet cell carcinoids and adenocarcinoma ex goblet cell carcinoids of the appendix is distinct from typical carcinoids and colorectal adenocarcinomas
Modern Pathology (2018)
-
Clinicopathologic and molecular analysis of disseminated appendiceal mucinous neoplasms: identification of factors predicting survival and proposed criteria for a three-tiered assessment of tumor grade
Modern Pathology (2014)
-
Becherzellkarzinoid des Rektums
Der Pathologe (2013)
-
Gastrointestinale gemischte adenoneuroendokrine Karzinome
Der Pathologe (2012)
-
Goblet cell carcinoid of the rectum with lymph node metastasis: Report of a case
Surgery Today (2011)
