
Abstract
There are approximately 200,000 new cases of cutaneous squamous cell carcinoma diagnosed each year in the United States, with between 1300 and 2300 deaths per year from metastatic disease. The tumor suppressor p16, encoded by the CDKN2/INK4a locus, has been reported mutated in ≥24% of squamous cell carcinomas. Mutations of the p16 gene have also been found in actinic keratoses, the first identifiable lesion in the continuum from normal skin to squamous cell carcinoma. We hypothesized that there may be an appreciable difference in expression of p16 between normal skin, actinic keratoses, squamous cell carcinoma in situ, and invasive squamous cell carcinoma. Ten actinic keratoses, 10 in situ squamous cell carcinomas, and 10 invasive squamous cell carcinomas were examined using the immunoperoxidase method with antigen retrieval for anti-p16INK4a antibody. All 10 actinic keratoses were positive for weak to moderate p16 staining in the lower third to lower half of the epidermis (especially the basal keratinocytes). This staining was significant when compared with the lack of staining seen in normal skin controls. Twenty percent of in situ squamous cell carcinomas had moderate to strong staining in only the lower half to lower two thirds of the epidermis, whereas 70% of the in situ squamous cell carcinomas exhibited full-thickness p16 staining, with no staining in the dermis. Thirty percent of invasive squamous cell carcinomas had full-thickness staining of the in situ component of the lesion, and 100% of invasive squamous cell carcinomas exhibited moderate to strong staining of the invasive component of the lesion. These findings indicate correlation between the increased expression of p16 during the progression of skin from actinic keratosis to in situ squamous cell carcinoma to invasive squamous cell carcinoma. These data may lend further support to the view of the actinic keratosis as a precursor lesion to squamous cell carcinoma.
Similar content being viewed by others


INTRODUCTION
Squamous cell carcinomas of the skin are one of the most common tumors. Approximately 200,000 cases of squamous cell carcinoma are diagnosed each year in the United States and cause between 1300–2300 deaths per year (1). The most common genetic abnormality of squamous cell carcinomas, seen in approximately 80%, are UV-type mutations of p53, a tumor suppressor known to induce apoptosis (2). Restriction point control of the cell cycle is another area of dysfunction found in squamous cell carcinomas. The retinoblastoma (Rb) gene product is the key gatekeeper regulating the passage of the cell through the G1/S restriction point of the cell division cycle. A dysfunction in the Rb pathway can lead to premature cell cycle progression, before DNA repair is completed, allowing genetically flawed cells to divide (3). The release of the Rb blockade is dependent on the enzyme activity of the molecular complex of cyclin-dependent kinase 4/6 (CDK4/6) and cyclin D (4). This complex normally phosphorylates and inactivates Rb to allow DNA replication to begin.
p16INK4a (p16) is a tumor suppressor protein that competes with cyclin D for binding to CDK4/6 (5). p16 is encoded by the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, also known as multiple tumor suppressor 1 (MTS1), located on chromosome 9p21 (6). The inactivity of the p16-CDK4/6 complex allows the active Rb to continue its arrest of the cell cycle. If the p16-CDK4/6 complex fails to form because of inadequate or incorrect expression of p16, cell division can proceed with the potential propagation of genetically flawed cells.
p16INK4a gene mutations have been found in ≤24% of squamous cell carcinomas (7, 8); however, expression of p16 has not been evaluated in cutaneous squamous cell carcinomas. Interestingly, Pavey et al. (9, 10) has shown that UV radiation induces p16 expression in normal skin and causes cell cycle arrest without p53 involvement. This raises the question as to the possible changes in p16 expression that may be seen as the skin, damaged by UV radiation, progresses in the putative pathway from normal skin to invasive squamous cell carcinoma. Therefore, the purpose of this study was to evaluate p16 expression using immunohistochemical techniques on known in situ and invasive squamous cell carcinomas arising in the setting of sun-damaged skin and their precursor lesion, the actinic keratosis.
MATERIALS AND METHODS
Immunohistochemistry
Formalin-fixed, paraffin-embedded specimens of normal skin, actinic keratoses, in situ squamous cell carcinomas, and invasive squamous cell carcinomas were randomly obtained from the University of Arkansas for Medical Sciences archive. All cases were reexamined by a board-certified dermatopathologist (B.R.S.) to confirm the histologic diagnosis. Four-micrometer recut sections were deparaffinized and rehydrated through a graded series of ethanol. The sections were pressure-cooked at 20 psi for 10 minutes in DAKO target retrieval solution (DAKO, Carpinteria, CA). After cooling to 37° C and rinsing in PBS, endogenous peroxidases were blocked with DAKO blocking solution (H2O2 and NaN3, DAKO, S2001) for 10 minutes. Nonspecific proteins were blocked by 30-minute incubation with DAKO protein block serum free (DAKO, X0909). Between each step, the sections were rinsed in PBS.
The sections were then incubated for 60 minutes with PharMingen p16 antibody G175–405 (550834, monoclonal mouse anti-human p16 INK4 IgG, BD Biosciences PharMingen, San Diego, CA) at 1:25 dilution. PharMingen polyclonal, biotin-conjugated, goat, anti-mouse IgG1, k (PharMingen 550337) diluted 1:50 was used as the secondary antibody followed by PharMingen Streptavidin-HRP (PharMingen 550946), both incubated for 30 minutes each. Reaction products were visualized with Biopath Bio-3,3′-diaminobenzidine chromagen (Biopath, Oklahoma City, OK) for 10 minutes, followed by counterstaining with hematoxylin and cover slipping.
Evaluation of p16 Expression
There were a total of 30 samples collected and analyzed, consisting of 10 actinic keratoses, 10 in situ squamous cell carcinomas, and 10 invasive squamous cell carcinomas. Eccrine gland and hair matrix cells served as internal positive controls, whereas lymphocytes served as internal negative controls. Normal skin specimen were also used as negative controls for keratinocyte staining. The intensity of chromagen staining was evaluated with the following grading system: 0, no staining; 1, weak staining; 2, moderate staining; and 3, strong staining.
RESULTS
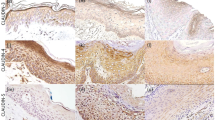
All 10 actinic keratoses examined were positive for weak to moderate staining in the lower third to lower half of the epidermis (Table 1). In several specimens, the staining was distinctly confined to the basal layer of keratinocytes. This staining was significant when compared with the lack of staining seen in normal skin control (Fig. 1A–B). Interestingly, the areas of acantholysis, corresponding presumably to the more atypical cells, exhibited the most intense staining.

A, high-power view of normal skin demonstrating absence of immunohistochemical staining for p16. B, high-power view of an actinic keratosis, demonstrating moderate staining for p16 along the basal layer of keratinocytes. The more intense staining is seen in the bases of the epidermal ridges.
Twenty percent of the in situ squamous cell carcinomas had moderate to strong staining in the only lower half to lower two thirds of the epidermis, whereas 70% of the in situ squamous cell carcinomas exhibited full-thickness staining, with no staining in the dermis (Table 1; Fig. 2A–B). One in situ squamous cell carcinoma exhibited only random staining similar to that seen in the invasive squamous cell carcinomas. Thirty percent of the invasive squamous cell carcinomas had full-thickness staining of the in situ component of the lesion, and 100% of the invasive squamous cell carcinomas exhibited moderate to strong staining of the invasive component of the lesion (Fig. 3). The most intense staining seen in the invasive squamous cell carcinomas was that of the cells invading the dermis. Interestingly, the invasive component of the lesions could be found by scanning for darkly staining areas within the dermis.

A, high-power view of squamous cell carcinoma in situ with strong, full-thickness, immunohistochemical staining for p16. B, low-power view of squamous cell carcinoma in situ showing the edge of the lesion. This demonstrates the strong full-thickness staining seen in the squamous cell carcinoma in situ in contrast to the virtual absence of staining of the surrounding normal skin.

High-power view of an area of dermal invasion in a squamous cell carcinoma demonstrating the scattered, moderate-to-strong staining for p16.
DISCUSSION
p16 inactivation by deletion, mutation, or methylation has been found in a wide range of human cancers, including breast, brain, skin, bladder, and prostate (11, 12). Seventy-five percent of melanoma cell lines have been found to contain mutant or lost p16 gene, and germ-line mutations have been found in the p16 gene in patients with multiple primary melanomas (13, 14). Funk et al. (15) found p16INK4a expression to be decreased in sporadic melanoma, and Keller-Melchoir et al. (16) found correlation between the loss of expression and the progression of melanoma from in situ to invasive to metastatic melanoma. However, there is controversy in the literature about p16 expression. Overexpression of p16 has been found in transformed cell lines from many tumor types when compared with their normal counterparts (17). Also, p16 expression has been shown to be up-regulated after UV irradiation of cultured melanocytes (18). This up-regulation is similar to what we have seen in the squamous cell carcinomas we evaluated in this study. This increased expression may be due to mutation in the CDKN2A gene with expression of inactive p16INK4a protein, resulting in the up-regulation of the gene product.
We also saw increased expression of p16 protein in 100% of the actinic keratoses that we examined. Actinic (or solar) keratoses are the thought to be either the precursor to or even the first identifiable changes of cutaneous squamous cell carcinoma. Actinic keratoses are usually skin-colored macules or papules measuring 1 to 4 mm in diameter but may be pigmented, erythematous, and/or hyperkeratotic. Actinic keratoses appear on the sun-damaged skin of Caucasians >40 years of age. The American Academy of Dermatology estimated that 60% of predisposed persons >40 years of age have at least one actinic keratosis (19). Although it is controversial as to what percentage of actinic keratoses will ultimately become invasive squamous cell carcinoma, like the squamous cell carcinoma, actinic keratoses are characterized histologically by keratinocyte atypia. Actinic keratoses not only share with squamous cell carcinomas cytological atypia, increased mitotic figures, and nuclear pleomorphism but also share the genetic hallmarks of cancer. Actinic keratoses share the same tumor markers and identical p53 gene mutations as squamous cell carcinoma (20). High frequency of loss of heterozygosity has been found in actinic keratoses, with 20% of actinic keratoses showing loss of eight or more alleles, 39% of those alleles from 9p (21). In fact, Soufir et al. (8) reported mutation of the p16 itself in an actinic keratosis, of the UVB signature type. And as discussed in the introduction, Pavey et al. (9, 10) reported that UV radiation induces p16 expression in normal squamous cells. This supports what we have seen in our study when we looked at the expression of p16 in actinic keratoses. The increased expression may be a protective mechanism to prevent the propagation of abnormal cells injured by UV radiation. The increased expression may also represent the overexpression of abnormal protein from mutation of the gene. This is supported by the more intense staining seen in the acantholytic, presumably more atypical regions within some of the actinic keratoses we examined. Further studies eliciting the exact protein identified by the antibodies used are necessary to make this distinction.
Another observation we made in this study was that although all of the actinic keratoses exhibited increased expression only in the basal and suprabasal region of the epidermis and the majority of in situ squamous cell carcinomas had full-thickness overexpression, full-thickness staining was seen in only a minority of the invasive lesions. In the majority of the invasive lesions examined, no full-thickness epidermal component could be found. This raises the discussion of tumor progression in cutaneous squamous cell carcinomas. It appears as though actinic keratoses may represent a precursor lesion on the pathway to the development of invasive squamous cell carcinomas. In contrast, in situ squamous cell carcinomas occurring in the setting of sun-damaged skin may be developing along a different pathway, with a different pattern of aberrant oncogene expression.
In conclusion, our study demonstrates a clear increase in p16 expression that correlates with the progression from normal skin to squamous cell carcinoma. Correlation was also seen in the differential staining within lesions between the more atypical areas of the actinic keratoses and the invasive squamous cells. This study lends further support to the view of actinic keratoses as an early identifiable stage in the development of squamous cell carcinoma. As most actinic keratoses do not inevitably progress to in situ or invasive squamous cell carcinoma, p16 overexpression appears to be necessary but not sufficient for this tumor progression, and other factors must be involved in this progressive transformation. Although this is a small study, these findings at least indicate the need for further study of the role of p16 in the pathway to cutaneous squamous cell carcinoma.
References
Miller DL, Weinstock MA . Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol 1994; 30: 774–778.
Bassett-Seguin N, Moles JP, Mils V . J Invest Dermatol 1994; 103: 102–106.
Sherr CJ . Cancer cell cycles. Science 1996; 274: 1672–1677.
Russo A, Tong L, Lee J, Jeffrey PD, Pavletich NP . Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumor suppressor p16INK4a. Nature 1998; 395: 237–243.
Serrano M . A new regulatory motif in cell-cycle control causing inhibition of cyclin D/CDK4. Nature 1993; 366: 704–707.
Kamb A, Shattuck-Eidens D . Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet 1994; 8: 23–26.
Kubo Y, Urano Y, Matsumoto K, Ahsan K, Arase S . Mutations of the INK4a locus in squamous cell carcinomas of human skin. Biochem Biophys Res Commun 1997; 232: 38–41.
Soufir N, Moles JP, Vilmer C, Moch C, Verola O, Rivet J, et al. p16 UV mutations in human skin epithelial tumors. Oncogene 1999; 18: 5477–5481.
Pavey S, Conroy S, Russel T, Gabrielli B . Ultraviolet radiation induces p16CDKN2A expression in human skin. Cancer Res 1999; 59: 4185–4189.
Pavey S, Russell T, Gabrielli B . G2 phase cell cycle arrest in human skin following UV irradiation. Oncogene 2001; 20: 6103–6110.
Nabori T, Miura K, Wu DJ, Lois A, et al. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994; 368: 753–756.
Raus M, Peters G . The p16INK4a/CDKN2A tumor suppressor and its relatives [review]. Biochim Biophys Acta 1998; 1378: 115–177.
Kamb A, Gruis NA . A cell cycle regulator potentially involved in genesis of many tumor types [report]. Science 1994; 264: 436–440.
Monzoon J, Liu L, Brill H, et al. CDKN2A mutations in multiple primary melanomas. N Engl J Med 1998; 338: 879–887.
Funk JO, Schiller PI, Barrett MT, Wong DJ, Kind P, Sander CA . p16INK4a expression is frequently decreased and associated with 9p21 loss of heterozygosity in sporadic melanoma. J Cutan Pathol 1998; 25: 291–296.
Keller-Melchoir R, Schmidt R, Piepkorn M . Expression of the tumor suppressor gene product p16INK4 in benign and malignant melanocytic lesions. J Invest Dermatol 1998; 110: 932–938.
Tam SW, Shay JW, Pagano M . Differential expression and cell cycle regulation of the cyclin-dependent kinase 4 inhibitor p16Ink4. Cancer Res 1994; 54: 5816–5820.
Piepkorn M . The expression of p16INK4a, the product of a tumor suppressor gene for melanoma, is upregulated in human melanocytes by UVB irradiation. J Am Acad Dermatol 2000; 42: 741–745.
Drake LA, Ceilley RI, Cornelison RL . Guidelines of care for actinic keratosis. J Am Acad Dermatol 1995; 32: 95–98.
Zeigler A, Jonason AS, Leffell DJ . Sunburn and p53 in the onset of skin cancer. Nature 1994; 372: 773–776.
Rehman I, Takata M, Wu YY, Rees JL . Genetic change in actinic keratoses. Oncogene 1996; 12: 2483–2490.
Acknowledgements
We acknowledge Vicky Givens and Lori Talley for their contributions in the lab.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hodges, A., Smoller, B. Immunohistochemical Comparison of P16 Expression in Actinic Keratoses and Squamous Cell Carcinomas of the Skin. Mod Pathol 15, 1121–1125 (2002). https://doi.org/10.1097/01.MP.0000032536.48264.D1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000032536.48264.D1
Keywords
This article is cited by
-
Expression of Ki-67 and P16 are related with HPV in squamous cell carcinoma of the external auditory canal
Journal of Otolaryngology - Head & Neck Surgery (2022)
-
Modeling transcriptomic age using knowledge-primed artificial neural networks
npj Aging and Mechanisms of Disease (2021)
-
Evidence for a non-stochastic two-field hypothesis for persistent skin cancer risk
Scientific Reports (2020)
-
Comparative study of p16 protein expression in squamous cell carcinomas from patients with epidermodysplasia verruciformis and patients without the disease
Archives of Dermatological Research (2017)
-
Human papillomavirus infection and p16 expression in the immunocompetent patients with extragenital/extraungual Bowen’s disease
Diagnostic Pathology (2016)
