
Abstract
The rhabdoid cell, which is typically observed in malignant rhabdoid tumor (MRT) and other malignant neoplasms, has an eosinophilic cytoplasm containing a spheroid perinuclear inclusion body. This distinct cell is known to act as a highly aggressive indicator in many types of malignant tumors and is characterized by aggregates of intermediate filaments, comprising both vimentin and cytokeratin (CK) 8, which is mainly expressed in simple-type epithelium such as liver and intestine. To clarify the cause of the inclusion body formation, we analyzed the alteration of the complete human CK8 gene (KRT 8: 1724 base pairs) in seven samples of MRT (three from frozen materials and four from cultured cell lines) by reverse-transcriptase polymerase chain reaction, followed by direct sequencing. In addition, the two cell lines, Huh7 and HeLa, which lacked rhabdoid feature, six pediatric malignant tumors, including three cases of primitive neuroectodermal tumor (PNET) and three of Wilms' tumor; and 15 normal liver tissue (as a control) were also analyzed. All MRT samples had missense mutations in the human KRT 8 gene, i.e., Arg89 → Cys (5/7); Arg → Cys251 (3/7); Glu267 → Lys (6/7); Ser290 → Ile, Met; (7/7) and Arg301 → His(4/7), none of which was detected in any control samples. Among these mutations, the most noteworthy findings were that Arg89 belongs to the H1 subdomain of the head domain and that Arg251 belongs to the short nonhelical linker segment, or L1–2. Both these mutations are noted for their relationships to lateral protofilament–protofilament interactions. In addition, Ser290 has been previously reported to be a phosphorylation site, which has been recognized to play an important role in filament organization, leading to conformational change of the CK8 filaments. In conclusion, mutated codons of CK8 gene in MRT were located in the important region involved in the conformational change of intermediate filament.
Similar content being viewed by others

Main
Malignant rhabdoid tumor (MRT) is extremely rare but is known for its highly aggressive malignancy in childhood and is characterized by the presence of a “rhabdoid cell” (1). The rhabdoid cell is regarded as an indicator of highly malignant potential in various types of specific malignant neoplasms, either mesenchymal or epithelial tumors, and numerous reports have indicated a relationship between this feature and poor prognosis (2, 3, 4, 5, 6, 7). This distinct cell has an eosinophilic cytoplasm containing a spheroid perinuclear inclusion body and is ultrastructurally characterized by the presence of aggregates of intermediate filaments of cytokeratin (CK) and vimentin (8). We have previously reported the details of CK expression in MRT, which were confined to mainly two subtypes of CKs, CK8 and CK18, in an analysis of a large panel of CK subtypes, and we have also clarified the subcellular distribution of CKs and vimentin in MRT cells (9, 10).
Intermediate filament forms a protein, 8 to 10 nm in diameter, that is the major cytoskeletal and nuclear matrix constituent of nearly all vertebrate cells, comprising a large family of cytoskeletal proteins including CKs. There are more than 20 different CK subtypes based on their sizes and isoelectric points (11, 12); these can be further subdivided into two subfamilies: acidic keratins, Type I (i.e., CK9–CK20) and neutral-basic keratins, Type II (i.e., CK1–CK8). CK filaments are obligatory heteropolymers that form heterotypic tetramer subunits containing two chains of representatives of each subfamily (i.e., CKs 8 and 18, CKs 5 and 14, and CKs 1 and 10). They exhibit a dense intermingled network extending from the nuclear periphery to the plasma membrane. Recently, by using molecular and cellular biological techniques, several skin-blistering diseases have been reported to be caused by CK gene mutations (CKs 5 and 14 and CKs 1 and 10) (13, 14, 15, 16, 17, 18, 19, 20, 21). Moreover, deleted or mutated CK filaments (CK8/18) in Escherichia coli have been found to cause the intracytoplasmic “inclusion body” (22). Although these discoveries have aroused renewed interest in the structural proteins of CKs, the function of CK8 has been rarely studied, and the precise mechanism of inclusion body formation remains unclear.
In this study, we investigated the alteration of the human CK8 (KRT 8) gene, which is one of the chief components of the inclusion body in MRT, and we attempted to ascertain the possible contribution of the KRT 8 gene to the formation of inclusion bodies.
MATERIALS AND METHODS
Clinical Samples and Cell Lines
As for clinical samples, all three frozen tissues, which had been previously diagnosed as MRT, were studied in our previous paper (23). Moreover, all cell lines have previously been described in published reports. The surgical specimens and cell lines of malignant rhabdoid tumors reported herein were obtained from the following institutions: Kyushu University, National Kyushu Medical Center, and Oita Red Cross Hospital. Dr. Ota (Shiga University of Medical Science) provided three MRT cell lines, Tm 87–16, STM91–01, and TTC549.
In summary, Tm 87–16 was established from pleural effusion of extrarenal MRT in a 21-month-old boy (24). STM91–01 was established from a pulmonary metastatic lesion of renal MRT in an 8-month-old boy (25). TTC549 was established from hepatic MRT in a 6-month-old girl (26), and we have recently established the cell line TC289, derived from renal MRT in a 1-year-old boy (27). In addition, as described elsewhere, six of the seven cases (85%) in this study revealed abnormalities of chromosome 22q11, which are well known as characteristic chromosomal abnormalities in MRT (27). The cell lines were cultured and maintained in RPMI-1640 supplemented with 10% fetal bovine serum in a humidified atmosphere of 5% CO2 and 95% air at 37°C. As other cell lines without any rhabdoid features, two famous tumor cell lines (HeLa and Huh7), which were known to express CK8, were also analyzed using the same method. Furthermore, for the purpose of excluding the base changes caused by polymorphism, 15 fresh nontumorous specimens of the liver tissue and 6 pediatric malignant tumors, including 3 cases of PNET (primitive neuroectodermal tumor) and 3 of Wilms' tumor, which expressed CK8 gene products, were also analyzed as control materials without any rhabdoid cells. These specimens were fresh enough to successfully analyze the sequence of the KRT 8 gene by reverse-transcriptase polymerase chain reaction (RT-PCR). All human samples were obtained under an institutional review board–approved protocol with subjects providing informed consent.
Electron Microscopic Study
The fresh samples were fixed in 3% glutaraldehyde solution (buffered pH, 7.4) and were postfixed in 1% phosphate-buffered osmium tetroxidate. After hydration, the tissue blocks were embedded in Epon 812 resin (TAAB Laboratories, Berks, UK) and cut on a Reichert ultramicrotome. Ultrathin sections were stained with uranyl acetate and lead citrate and examined under a JEM 100 C electron microscope (Jeol, Tokyo, Japan).
Immunohistochemical Study
We employed a previously reported method for the preparation of sections from cultured cells to cell blocks (28). Immunohistochemical studies were performed as previously described using the antibodies, comprising CK4 (ICN), CK6 (PROGEN), CK7 (DAKO), CK8 (DAKO), CK10 (ICN), CK13 (Boehringer), CK16 (NOVO), CK17 (DAKO), CK18 (DAKO), CK19 (DAKO), CK20 (DAKO), EAB903 (reacts with CK1, CK5, CK10 and CK14; Enzo), and AE5 (reacts with CK3, CK12, YLEM), by use of a streptavidin-biotin-peroxidase kit (Nichirei, Tokyo, Japan), with counterstaining by hematoxylin (9). The degree of immunohistochemical expression was classified as follows: diffusely positive (>50% of the cells were positive), focally positive (10 to 50% of the cells were positive), or negative (<10% of the cells were positive). The immunoreactivity was judged independently by the three pathologists (HS, SadT, and YuO) who were unaware of the results of other analyses. All the slides were independently reviewed twice, and those that were the subject of intraobserver disagreements were reviewed a third time, followed by a conclusive judgment.
Reverse Transcriptase–PolymeraseChain Reaction
The sequences of the primers and PCR conditions are summarized in Table 2. These six primer pairs were designed to cover all sequences of the human KRT 8 mRNA gene (GenBank Accession No. AAA35748: 483 residues, 1724 base pairs). Total RNA was extracted from each of the snap-frozen tumor tissue samples and cell lines using Trizol reagent (GIBCO, BRL, Gaitherburg, MD), according to the suggested procedures. Reverse transcription was performed with 5 μg of total RNA in a total volume of 20 μL according to the manufacturer's protocol. Single-stranded cDNA was amplified by PCR, with 10 nmol each of forward and reverse primers, by use of Ampli Taq Gold DNA polymerase (Perkin-Elmer). Denaturation (94°C for 1 min) and extension (72°C for 1 min) were performed according to standard methods.
Sequencing and Identification ofCytokeratin Mutations
We sequenced RT-PCR products after subcloning by using the dye-terminator method. After purification, direct sequencing was carried out by the dideoxy chain termination methods using a Perkin Elmer ABI Prism 310 sequence analyzer (Applied Biosystems, Foster City, CA). Both sense and antisense strands were sequenced for confirmation. In addition, to exclude the effect of PCR artifacts, total RNA was extracted from three different portions of each material, then we confirmed that these sequences were reproducible. Moreover, if the base substitutions were suggested, we confirmed that these substitutions were surely observed by use of two different primers.
RESULTS
Microscopic and Clinical Findings
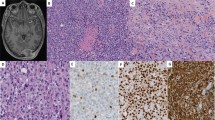
Individual cells were poorly differentiated and round or polygonal, with an elongated strap form. Characteristically, many of the tumor cells had eosinophilic glassy cytoplasm that contained large, spherical, hyalinelike inclusion bodies (Fig. 1A). These peculiar structures were located near the nuclei. The nuclei, which were only slightly irregular, round to oval, and vesicular, usually had one, large prominent nucleoli, with occasional mitosis. Four MRT cell lines microscopically inherited the character of the primary rhabdoid tumors. The other control cell lines and liver tissue did not show any rhabdoid features. The clinical findings are summarized in Table 1.

A, histologic features of malignant rhabdoid tumor. A solid nest of rounded or polygonal cells with eccentric nuclei and prominent nucleoli. Glassy eosinophilic cytoplasm containing hyalin-like inclusion bodies is evident (Case 1, hematoxylin and eosin stain; original magnification, 500×). B, electron microscopic feature. Higher magnification of aggregates of intermediate filaments in a paranuclear location (magnification, 18,000×). C, the tumor cells reveal immunoreactivity for both cytokeratin (CK) 8 and CK18, despite showing negative immunoreactivity for other types of CK.
Ultrastructural Findings
The majority of the tumor cells were oval or polygonal, with a minority of spindle-shaped cells. Both clinical cases and cell line materials showed similar findings with irregular nuclei that were often compressed and eccentrically located. The striking cytoplasmic feature of the tumor cells was the presence of bundles of cytoplasmic filaments, often occupying paranuclear areas (Fig. 1B). The filaments were approximately 10 nm in diameter and represented intermediate filaments. These filamentous inclusions varied in size and arrangement from cell to cell.
Immunohistochemical Findings
The immunohistochemical results are summarized in Table 1. All of the seven MRT samples in this study showed a diffusely positive immunoreactivity in the inclusion bodies for both CK8 and CK18 (7/7) while also showing focally positive immunoreactivity for CK10 (2/7) and CK13 (1/7; Fig. 1C). On the other hand, other CKs, including CK1, CK3, CK4, CK5, CK6, CK7, CK12, CK14, CK16, CK17, CK19, and CK20, were completely negative in all samples. Vimentin expression was found in all samples. HeLa, Huh7, and controlled liver tissue also showed positive immunoreactivity for CKs 8 and 18 but no detectable inclusion bodies.
Mutation Analysis of CK8 in MRT and Other Control Materials
RT-PCR revealed KRT8 gene products in all cases and cell lines, including controlled materials. Overall, the human KRT 8 base substitutions were detected in all three MRT frozen materials as well as in four MRT cell lines. Table 3 summarizes the mutational data of all samples. We identified missense mutations at Codon 89 (5 cases: CGC → TGC [Arg → Cys]), Codon 251 (3 cases: CGC → TGC [Arg → Cys]), Codon 267 (6 cases: GAG → AAG [Glu → Lys]), Codon 290 (4 cases: AGC → ATC [Ser → Ile]; 3 cases: AGC → ATG [Ser → Met]), and Codon 301 (4 cases: CGC → CAC [Arg → His]; Fig. 2A). Arg89 belongs to the H1 subdomain of the head domain, and Arg251 belongs to the short nonhelical linker segment or L1–2 (Fig. 3). In addition, Ser290 has been previously reported as a phosphorylation site of the rod domain. These mutations were not detected in any of 15 controlled materials of the liver tissue, in six controlled pediatric malignancies, or in two other cell lines. In addition, five silent mutations were also detected: Codon 190 (6 cases: AAC → AAT [Asn → Asn]), Codon 217 (7 cases: ACC → ACT [Thr → Thr]), Codon 259 (4 cases: ATT → ATC [Ile → Ile]), Codon 269 (5 cases: ATT → ATC [Ile → Ile]), and Codon 307 (7 cases: TCA → TCT [Ser → Ser]). Fifteen controlled samples and two cell lines also contained some of the above silent mutations (Fig. 2B).

Reverse-transcriptase polymerase chain reaction and direct sequence analysis at Codons 89, 251, and 290 of the human KRT 8 gene in Case 2 and the control liver tissue. The base sequences of 15 control tissues in the present region were completely in accord with those in GenBank. C to T transition was detected at Codon 89, C to T transition at Codon 251, and G to T transversion at Codon 290 (A). For an evaluation of base sequencing, 15 control samples and two famous cell lines without rhabdoid feature were also studied. We identified missense mutations in four of the seven malignant rhabdoid tumor (MRT) cases at Codon 301 (arrow, CGC → CAC). On the other hand, we identified silent mutations in all control samples as well as seven MRT cases at Codon 307 (arrow, TCA → TCT). In addition, “CK8RT4” shows the base sequences in GenBank (Accession No. AAA35748; B).

Diagrammatic representation of cytokeratin (CK) 8 to show the positions of gene mutation observed in malignant rhabdoid tumor (MRT), relative to the previously reported CK5 (KRT 5) gene mutations in patients affected by Weber-Cockayne epidermolysis bullosa simplex (EBS-WC) or epidermolytic hyperkeratosis (EH) patients. Stick figure depicts secondary structure of human CKs. The large boxes indicate the four helical subdomains (1A, 1B, 2A, and 2B), and the shaded boxes denote the highly conserved end domains of the rod. Solid bars denote nonhelical head and tail domains, and the head domain contains a highly conserved H1 subdomain. In the H1 subdomain (white box), the end domains of the rod 1A and the L1–2 region between 1B and 2A are expanded to show the amino acid sequence alignment of Type II CKs. The CK1 (KRT 1) gene mutation observed in a patient with EH, the CK5 (KRT 5) gene mutations observed in patients with EBS-WC, and the CK8 (KRT 8) gene mutations observed in this study are also shown. Note the CK8 gene mutations relative to the H1 and L1–2 regions and relative to known CK point mutations in EBS-WC and EH. The Arg (R) 251 → Cys (C) mutation in the CK8gene is located in the L1–2 domain, as are the Met (M) 325 → Thr (T), the Asn (N) 327 → Lys (K), and the Arg (R) 331 → Cys (C) mutations in the CK5 gene of the EBS-WC families (15, 16, 18). MRTCK8 shows the CK8 missense mutation sites observed in this study. The Arg (R) 89 → Cys (C) mutation in the CK8 gene is located in the H1 subdomain, as is the Ile (I) 161 → Ser (S) mutation in the CK5 gene of the EBS-WC families (17) and the Leu (L) 149 → Pro (P) mutation in the CK1 gene of EH (14).
DISCUSSION
Cytokeratins (CKs) 8 and 18, which are expressed preferably in simple-type epithelium such as liver, intestine, and pancreas, are discussed in connection with hepatocellular degeneration, in particular chronic hepatitis, liver cirrhosis (29), and Mallory bodies (30). With regard to the human KRT 18 gene, a few mutagenic residues have been described in limited human diseases. For example, previous studies demonstrated the KRT 18 gene mutation in cryptogenic cirrhosis and chronic hepatitis (29, 31, 32). On the other hand, the human KRT 8 gene, located in chromosome 12q13, has been described in several reports regarding genetic sequence and the regulation of mRNA (33, 34); however, to our knowledge, there have been no reports of the human KRT 8 gene mutation in neoplasms. In addition, few chromosomal abnormalities of MRT have been demonstrated, other than hSNF5/INI1, which maps to chromosome 22q11 (26, 35, 36, 37, 38, 39). In this study, we described for the first time the KRT 8 gene mutation in MRT, which is characterized by the intracytoplasmic inclusion body of CK filaments.
Similar to all other intermediate filaments, CKs have the characteristic structural features of a central amino acid: a coiled, α-helical domain that is flanked by NH2− and COOH− terminal globular head and tail domains, respectively. Although the mechanisms regulating intracellular assembly or disassembly of CK filaments have been obscure, recent analysis of mutant CK proteins has suggested that at least three important regions related to assembly into a filament structure are involved in lateral protofilament–protofilament interactions. These three regions are as follows: the H1 subdomain of the head domain and the beginning of the α-helical domain; the short nonhelical linker segment, or L1–2; and some major sites for phosphorylation located mainly in the head and tail domains. We noted that the five base substitutions (Codons 89, 251, 267, 290, and 301) were not observed in any of 15 control samples and were observed only in the MRT samples. The most striking finding in this study is that all of these mutated codons were located in the above three important regions.
First of all, hot mutation spots in some skin-blistering diseases with pairs CKs 5 and 14 or CKs 1 and 10 involve the H1 subdomain of the head domain and the beginning of the rod 1A domain of CKs (20, 21). Recently, these mutation spots of the human KRT 18 gene have been reported in the transgenic mouse model that develops chronic hepatitis (29). In particular, the H1 subdomain of the head influenced the CK assembly of skin diseases (14, 17, 40) and HeLa cells (41); thus, the region has been commonly assumed to play an essential role in the alignment of CK filaments. Actually, the mild aggregation and waviness of basal-cell CK filaments were observed in some skin-blistering diseases (17). The same region has been reported to be important for correct filament polymerization in CK8 (42). Figure 3 indicates a list of amino acid sequences for the H1 subdomain from the available Type II CKs obtained from GenBank, and this region shows a very highly conserved sequence within Type II CKs. The strict role of the H1 subdomain is presently unknown; however, a temporal relationship between mutation in the H1 region and alterations in phosphorylation of the regional proteins has been noted (17). Thus, the mutation in the H1 subdomain could be one of the candidates that can explain the formation of abnormal CK filaments. In the current study, the Arg89 → Cys mutation (5/7) was located in the H1 subdomain.
Second, the other important region for CK assembly is the short nonhelical linker segment, or L1–2. We identified the Arg251 → Cys mutation in the L1–2 region (3/7). Figure 3 indicates a list of normal amino acid sequences for L1–2, which are conserved among Type II CKs. Mutation of the Arg331 of the human KRT 5 gene, which can be conservatively substituted by Arg251 of the human KRT 8 gene, has been reported in skin-blistering disease, such as Weber-Cockayne epidermolysis bullosa simplex (EBS-WC; 16). Other investigators have reported L1–2 mutations of Met 327 and Asn329 of the human KRT 5 gene in EBS-WC (18). The L1–2 segment is thought to play an important role in lateral interactions through the following mechanism: two staggered dimers are aligned so that the carboxy half of the L1–2 domain in one dimer is in close proximity to, and possibly even directly opposite from, the helix 1A and head H1 segment in an adjacent dimer (43). Thus, it is possible that this Arg251 mutation is also involved in lateral protofilament–protofilament interactions, in cooperation with the mutation of the above-mentioned H1 subdomain, seen as Arg89 → Cys.
Finally, the sequences in the head and tail domains have several phosphorylation sites, which can orchestrate dynamic IF assembly and disassembly events in vivo (44). The reported phosphorylation sites of CK8 are Codons 8, 12, 14, 23, 33, 36, 42, and 50 in the head and Codons 416, 423, 425, 431, 441, and 456 in the tail regions (45, 46). Other phosphorylation sites have also been reported in the rod (Codon 290; 46). It is possible that this Ser290 can also make a contribution to assembly into a filament structure. In this study, it is interesting that the Ser290 mutations to Met or Ile were identified in all MRT samples (7/7).
In addition, our results showed that a few silent mutations, differing from the KRT 8 gene sequence in GenBank, were identified in the control samples as well as in the MRT samples (Fig. 2B). We believe most of these substitutions were polymorphisms or racial difference, because the cytoskeletal molecules including the CK families were known to be rich in genetic polymorphism (47).
To summarize, we first demonstrated the human KRT 8 gene mutations in MRT. As a consequence of analyzing these mutations, all of the mutated codons were located in the important regions of keratin filaments, although these results provide no evidence that KRT 8 gene mutations themselves are directly relevant to conformational change of intermediate filaments, leading to the formation of inclusion bodies in MRT. Further examination of the function of CK filaments, using other molecular and cellular biological techniques, would clarify the morphological character of the inclusion bodies in MRT.
Accession codes
References
Beckwith JB, Palmer NF . Histopathology and prognosis of Wilms tumor. Results from the First National Wilms' Tumor Study. Cancer 1978; 41: 1937–1948.
Tsuneyoshi M, Daimaru Y, Hashimoto H, Enjoji M . The existence of rhabdoid cells in specified soft tissue sarcomas. Histopathological, ultrastructural and immunohistochemical evidence. Virchows Arch 1987; 411: 509–514.
Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S . Survival in synovial sarcoma. A multivariate study of prognostic factors with special emphasis on comparison between early death and long-term survival. Am J Surg Pathol 1993; 17: 35–44.
Ueyama T, Nagai E, Yao T, Tsuneyoshi M . Vimentin-positive gastric carcinomas with rhabdoid features. A clinicopathologic and immunohistochemical study. Am J Surg Pathol 1993; 17: 813–819.
Miettinen M, Fanburg-Smith JC, Virolainen M, Shmookler BM, Fetsch JF . Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of differential diagnosis. Hum Pathol 1999; 30: 934–942.
Oshiro Y, Shiratsuchi H, Oda Y, Toyoshima S, Tsuneyoshi M . Rhabdoid feature in leiomyosarcoma of soft tissue: with special reference to aggressive behavior. Mod Pathol 2000; 13: 1211–1218.
Oshiro Y, Shiratsuchi H, Tamiya S, Oda Y, Toyoshima S, Tsuneyoshi M . Extraskeletal myxoid chondrosarcoma with rhabdoid features, with special reference to its aggressive behavior. Int J Surg Pathol 2000; 8: 145–152.
Vogel AM, Gown AM, Caughlan GJ, Haas JE, Beckwith JB . Rhabdoid tumors of the kidney contain mesenchymal specific and epithelial specific intermediate filament proteins. Lab Invest 1984; 50: 232–238.
Shiratsuchi H, Oshiro Y, Saito T, Itakura E, Kinoshita Y, Tamiya S, et al. Cytokeratin subunits of inclusion bodies in rhabdoid cells: immunohistochemical and clinicopathological study of malignant rhabdoid tumor and epithelioid sarcoma. Int J Surg Pathol 2001; 9: 37–48.
Itakura E, Tamiya S, Morita K, Shiratsuchi H, Kinoshita Y, Oshiro Y, et al. Subcellular distribution of cytokeratin and vimentin in malignant rhabdoid tumor cells: three-dimensional imaging with confocal laser scanning microscopy and double immunofluorescence. Mod Pathol 2001; 14: 854–861.
Moll R, Franke WW, Schiller DL . The catalog of human cytokeratins: patterns of expression in normal epithelial, tumors and cultured cells. Cell 1982; 31: 11–24.
Moll R, Löwe A, Laufer J, Franke WW . Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am J Pathol 1992; 140: 427–447.
Ishida-Yamamoto A, McGrath JA, Chapman SJ, Leigh IM, Lane EB, Eady RAJ . Epidermolysis bullosa simplex (Dowling-Meara type) is genetic disease characterized by an abnormal keratin filament network involving keratins K5 and K14. J Invest Dermatol 1991; 97: 959–968.
Chipev CC, Korge BP, Morkova N, Bale SJ, DiGiovanna JJ, Compton JG, et al. A leucine → proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell 1992; 70: 821–828.
Humphries MM, Sheils DM, Farrar GJ, Kumar-Singh R, Kenna PF, Mansergh FC, et al. A mutation (Met → Arg) in the type I keratin (K14) gene responsible for autosomal dominant epidermolysis bullosa simplex. Hum Mutat 1993; 2: 37–42.
Rugg EL, Morley SM, Boxer M, Tidmen MJ, Navsaria H, Leigh IM, et al. Missing links: Weber-Cockayne keratin mutations implicate the L12 linker domain in effective cytoskeleton function. Nat Genet 1993; 5: 294–300.
Chan YM, Yu QC, Fine JD, Fuchs E . The genetic basis of Weber-Cockayne epidermolysis bullosa simplex. Proc Natl Acad Sci U S A 1993; 90: 7414–7418.
Chan YM, Yu QC, LeBlanc-Straceski J, Christiano A, Pulkkinen L, Kucherlapati RS, et al. Mutations in the non-helical linker segment L1–2 of keratin 5 in patients with Weber-Cockayne epidermolysis bullosa simplex. J Cell Sci 1994; 107: 765–774.
Fuchs E . Intermediate filaments and disease: mutations that cripple cell length. J Cell Biol 1994; 125: 511–516.
Yang JM, Chipev CC, DiGiovanna JJ, Bale SJ, Marekov LN, Steinert PM, et al. Mutations in the H1 and 1A domains in the keratin 1 gene in epidermolytic hyperkeratosis. J Invest Dermatol 1994; 102: 17–23.
McLean WHI, Eady RAJ, Dopping-Hepenstal PJC, McMillan JR, Leigh IM, Navsaria HA, et al. Mutations in the rod 1A domain of keratins 1 and 10 in bullous congenital ichthyosiform erythroderma (BCIE). J Invest Dermatol 1994; 102: 24–30.
Magin TM, Hatzfeld M, Franke WW . Analysis of cytokeratin domains by cloning and expression of intact and deleted polypeptides in Escherichia coli. EMBO J 1987; 6: 2607–2615.
Kinoshita Y, Shiratsuchi H, Tamiya S, Oshiro Y, Oda Y, Suita S, et al. Mutations of the p53 gene in malignant rhabdoid tumors of soft part and the kidney: immunohistochemical and DNA direct sequencing analysis. J Cancer Res Clin Oncol 2001; 127: 351–358.
Karnes PS, Tran TN, Cui MY, Bogenmann E, Shimada H, Ying KL . Establishment of rhabdoid tumor cell line with a special chromosomal abnormality, 46, XY, t(11;22)(p15.5;q11.23). Cancer Genet Cytogenet 1991; 56: 31–38.
Ota S, Crabbe DCG, Tran TN, Triche TJ, Shimada H . Malignant rhabdoid tumor. A study with two established cell lines. Cancer 1993; 71: 2862–2872.
Biegel JA, Allen CS, Kawasaki K, Shimizu N, Budarf ML, Bell CJ . Narrowing the critical region for a rhabdoid tumor locus in 22q11. Genes Chromosomes Cancer 1996; 16: 94–105.
Kinoshita Y, Tamiya S, Oda Y, Mimori K, Inoue H, Ohta S, et al. Establishment and characterization of malignant rhabdoid tumor of the kidney. Oncol Rep 2000; 8: 43–48.
Xu XC, Clifford JL, Hong WK, Lotan R . Detection of nuclear retinoic acid receptor mRNA in histological tissue sections using nonradioactive in situ hybridization histochemistry. Diagn Mol Pathol 1994; 3: 122–131.
Ku NO, Michie SA, Oshima RG, Omary MB . Chronic hepatitis, hepatocyte fragility, and increased soluble phosphoglycokeratins in transgenic mice expressing a keratin 18 conversed arginine mutant. J Cell Biol 1995; 131: 1303–1314.
Salmhofer H, Rainer I, Zatloukal K, Denk H . Posttranslational events in griseofulvin-induced keratin cytoskeleton alterations. Hepatology 1994; 20: 731–740.
Ku NO, Michie SA, Soetikno RM, Resurreccion EZ, Broome RL, Oshima RG, et al. Susceptibility to hepatotoxicity in transgenic mice that express a dominant-negative human keratin 18 mutant. J Clin Invest 1996; 98: 1034–1046.
Ku NO, Wright TL, Terrault NA, Gish R, Omary MB . Mutation of human keratin 18 in associated with cryptogenic cirrhosis. J Clin Invest 1997; 99: 19–23.
Yamamoto R, Kao LC, Mcknight CE, Strauss JF III . Cloning and sequence of cDNA for human placental cytokeratin 8. Regulation of mRNA in trophoblastic cells by cAMP. Mol Endocrinol 1990; 4: 370–374.
Krauss S, Franke WW . Organization and sequence of the human gene encoding cytokeratin 8. Gene 1990; 86: 241–249.
Besnard-Guerin C, Cavenee W, Newsham I . The t(11;22)(p15.5;q11.23) in a retroperitoneal rhabdoid tumor also includes a regional deletion distal to CRYBB2 on 22q. Genes Chromosomes Cancer 1995; 13: 145–150.
Schofield DE, Beckwith JB, Sklar J . Loss of heterozygosity at chromosome regions 22q11–12 and 11p15.5 in renal rhabdoid tumors. Genes Chromosomes Cancer 1996; 15: 10–17.
Rosty C, Peter M, Zucman J, Validire P, Delattre O, Aurias A . Cytogenic and molecular analysis of a t(1;22)(p36;11.2) in a rhabdoid tumor with a putative homozygous deletion of chromosome 22. Genes Chromosomes Cancer 1998; 21: 82–89.
DeCristofaro MF, Betz BL, Wang W, Weissman BE . Alteration of hSNF5/INI1/BAF47 detected in rhabdoid cell lines and primary rhabdomyosarcomas but not Wilms' tumors. Oncogene 1999; 18: 7559–7565.
Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998; 394: 203–206.
Steinert PM, Parry DAD . The conserved H1 domain of the type II keratin 1 chain plays an essential role in the alignment of nearest neighbor molecules in mouse and human keratin 1/keratin 10 intermediate filaments and the two- to four-molecule level of structure. J Biol Chem 1993; 268: 2878–2887.
Chen PH, Ornelles DA, Shenk T . The adenovirus L3 23-kilodalton proteinase cleaves the amino-terminal head domain from cytokeratin 18 and disrupts the cytokeratin network of HeLa cells. J Virol 1993; 67: 3507–3514.
Böttger V, Lane B . A monoclonal antibody epitope on keratin 8 identified using a phage peptide library. J Mol Biol 1994; 235: 61–67.
Steinert PM, Marekov LN, Fraser RDB, Parry DAD . Keratin intermediate filament structure. Crosslinking studies yield quantitative information on molecular dimensions and mechanism of assembly. J Mol Biol 1993; 230: 436–452.
Busso N, Musur SK, Lazega D, Waxman S, Ossowski L . Induction of cell migration by pro-urokinase binding to its receptor: possible mechanism for signal transduction in human epithelial cells. J Cell Biol 1994; 126: 259–270.
Ando S, Tokui T, Yano T, Inagaki M . Keratin 8 phosphorylation in vitro by cAMP-dependent protein kinase occurs within the amino- and carboxyl-terminal end domains. Biochem Biophys Res Commun 1996; 221: 67–71.
Ku NO, Omary MB . Phosphorylation of human keratin 8 in vivo at conserved head domain serine 23 and at epidermal growth factor-stimulated tail domain serine 431. J Biol Chem 1997; 272: 7556–7564.
Sorensen CB, Ladekjaer-Mikkelsen AS, Andresen BS, Brandrup F, Veien NK, Buus SK, et al. Identification of novel and known mutations in the genes for keratin 5 and 14 in Danish patients with epidermolysis bullosa simplex: correlation between genotype and phenotype. J Invest Dermatol 1999; 112: 184–190.
Acknowledgements
The authors sincerely thank Kyushu University, National Kyushu Medical Center, and Oita Red Cross Hospital for contributing their cases to this study. We also thank Dr. Ota (Shiga University of Medical Science) for his kind gift of three MRT cell lines. We also thank Dr. Y. Hachitanda (Kyushu University) and Dr. K. Arima (Saga Medical School) for their fruitful comments, Miss T. Ishikawa for her excellent practical assistance (Kyushu University), and Miss Katherine Miller (Royal English Language Center, Fukuoka, Japan) for revising the English used in this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported in part by the Fukuoka Anticancer Society and by a Grant-in-Aid for Scientific Research (No. 12670167) from the Ministry of Education, Science and Culture, Japan.
Rights and permissions
About this article
Cite this article
Shiratsuchi, H., Saito, T., Sakamoto, A. et al. Mutation Analysis of Human Cytokeratin 8 Gene in Malignant Rhabdoid Tumor: A Possible Association with Intracytoplasmic Inclusion Body Formation. Mod Pathol 15, 146–153 (2002). https://doi.org/10.1038/modpathol.3880506
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880506
Keywords
This article is cited by
-
Malignant Rhabdoid Tumor of the Floor of Mouth: First Reported Case in the Oral Cavity of an Adult
Head and Neck Pathology (2015)
-
Improved survival in patients with breast rhabdoid tumors with multi-agent adjuvant chemotherapy combined with irradiation
Cancer Chemotherapy and Pharmacology (2009)
-
Clone-specific expression, transcriptional regulation, and action of interleukin-6 in human colon carcinoma cells
BMC Cancer (2008)
-
Highly aggressive behavior of malignant rhabdoid tumor: a special reference to SMARCB1/INI1 gene alterations using molecular genetic analysis including quantitative real-time PCR
Journal of Cancer Research and Clinical Oncology (2007)
-
INI1 expression is retained in composite rhabdoid tumors, including rhabdoid meningiomas
Modern Pathology (2005)
