
Abstract
Renal schwannomas are extraordinarily rare neoplasms; only six have been reported, the majority of which occurred in the renal pelvis. We report the clinical and pathologic features of four additional cases. The resected kidney in all patients contained a well-demarcated, yellow-tan, smooth, and bulging intraparenchymal tumor (mean size, 9.7 cm; range, 4 to 16 cm). Microscopically, three cases were classified as cellular schwannomas, and one was a usual-type schwannoma, with degenerative nuclear atypia. By immunohistochemistry, all tumors were strongly S-100 protein positive and negative for pan-cytokeratin, CD57, smooth muscle actin, desmin, and CD34. Epithelial elements were not noted in the tumors, and there was no history of any clinical syndromes in these patients. Analysis of the four cases showed the mean age at presentation to be 47 years (range, 18 to 84 years), with no sex predisposition (two men, two women). Most patients were asymptomatic, and all received a diagnosis of renal cell carcinoma and treated as having such. Recognition and awareness of these rare, benign tumors will assist in the differential diagnosis of spindle cell tumors of the kidney and prevent their misdiagnosis as sarcomatoid carcinomas of the kidney or renal sarcomas. Our study, the largest series to date of renal schwannomas, demonstrates a predilection for the cellular variant in the kidney, documents that these tumors may present in the nonhilar region of the kidney, and provides clinical evidence of their benign biologic behavior.
Similar content being viewed by others

Main
Schwannoma, or neurilemoma, is a benign neoplasm of Schwann cells. The majority of schwannomas arise as solitary masses of either superficial or deep soft tissues, often in association with a nerve.(1, 2) Favored sites include cranial and spinal nerve roots, cervical nerves, and nerves in the flexor compartments of the forearm and lower leg.(1, 2) Although schwannomas arise not uncommonly in the retroperitoneum, usually in association with the lumbosacral nervous plexus, involvement of retroperitoneal parenchymal organs is very uncommon. In particular, schwannomas of renal origin are very rare, with only six reported cases.(3, 4, 5, 6, 7, 8, 9, 10) We report herein the clinicopathologic features of four additional cases of renal schwannoma.
METHODS
All of the available hematoxylin and eosin–stained sections were reviewed for each case. For immunohistochemistry, deparaffinized sections were immunostained with antibodies to S-100 protein (polyclonal, prediluted, Ventana, Tucson, AZ), CD34 (QBEND; 1:640; PharMingen, San Diego, CA), smooth muscle actin (1A4; 1:2; Ventana), desmin (D33, prediluted; Ventana), pan-cytokeratin (AE1/AE3; 1:50; Dako, Carpinteria, CA), and CD57 (Leu7; 1:10; Becton-Dickinson, Mountain View, CA). Negative controls consisted of substitutions of mouse ascites fluid for the primary antibody. Sections were subjected to heat-induced epitope retrieval, using a vegetable steamer. Antigens were localized using an avidin-biotin method with 3,3′-diaminobenzidine as a chromogen. All immunohistochemical studies were independently evaluated by two pathologists (IA-C, ALF). There were no disagreements in interpretation of these studies.
Gross Findings
The four tumors presented as well-circumscribed, intrarenal masses that compressed the adjacent renal parenchyma (Fig. 1). The tumors measured from 4 to 16 cm (mean, 9.7 cm) and were tan to yellow in color. Cystic change was noted in two cases, and one tumor contained scattered calcifications.

On gross pathologic examination, the tumors consisted of multinodular masses of yellow to tan tissue, with central softening or cystic change, and compression of the surrounding renal parenchyma.
Microscopic Findings
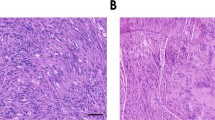
Case 1 showed the typical features of a schwannoma, with excellent circumscription; numerous thick-walled, hyalinized blood vessels; cellular Antoni A areas; and hypocellular Antoni B areas (Figs. 2 and 3). Nuclear palisading and Verocay body formation was prominent. The majority of the tumor cells were bland, uniform spindle cells with wavy nuclei. Scattered tumor cells showed nuclear enlargement with hyperchromasia and smudging of the chromatin. No mitotic activity or necrosis was present.

Low-power view of a typical schwannoma, with excellent circumscription, a dense fibrous capsule, and the absence of infiltration of the adjacent adrenal gland.

Antoni B area from a typical schwannoma. Note the hyalinized, ectatic blood vessel.
Cases 2 to 4 displayed the histologic features of cellular schwannomas. These tumors were characterized by excellent circumscription; fascicular growth; thick-walled, hyalinized blood vessels; cystic change with aggregates of foamy macrophages; and a mild lymphocytic infiltrate (Figs. 4 and 5). The tumors displayed relatively uniform cellularity, without distinct Antoni A and B areas, Verocay bodies, or nuclear palisading. All tumors had a very low mitotic rate (<1/10 high-power fields), and necrosis was not identified.

Three of the four cases were cellular schwannomas and were characterized by near-total Antoni A growth pattern, as illustrated here. Note the fascicular growth, the scattered lymphocytes, and the thick-walled blood vessel.

Aggregates of foamy macrophages may be a clue to the diagnosis of renal cellular schwannoma.
Immunohistochemical Findings
The fourcases were all strongly and uniformly positive with antibodies to S-100 protein (Fig. 6). No case expressed cytokeratin, CD34, pan-cytokeratin, CD57, smooth muscle actin, or desmin.

The four renal schwannomas were strongly positive for S-100 protein, as illustrated here.
DISCUSSION
In the adult, the majority of renal neoplasms are of epithelial origin. Mesenchymal renal tumors are far less common, with leiomyosarcoma the most common sarcoma and angiomyolipoma the most common benign mesenchymal neoplasm (11). Nerve sheath tumors of the kidney seem to be particularly rare, with only six previously reported schwannomas (3, 4, 5, 6, 7, 8, 9, 10), one reported neurofibroma (12), and four reported renal malignant peripheral nerve sheath tumors (13, 14, 15, 16). The clinicopathologic features of all reported renal schwannomas, including the current cases, are summarized in Table 1. Of these 10 tumors, 5 arose from the parenchyma and 3 arose from the hilus. In two cases, the clinical and pathologic information was not available (5, 6). Patients with retroperitoneal and renal schwannomas typically have nonspecific symptoms and signs. Malaise, weight loss, fever, and abdominal or flank pain are common findings (13, 14). A palpable abdominal mass is frequently present. Hematuria, a feature not previously described in renal schwannomas (3, 4, 5, 6, 7, 8, 9, 10), was present in one of our cases. An uneventful follow-up of 18 months was available in one of the six previously reported cases. Follow-up was available on all of the four cases reported herein (mean, 43 months; range, 12 to 60 months); all patients are disease-free.
Histopathologically, three of our four cases were classified as cellular schwannomas. Cellular schwannoma is a rare variant of schwannoma, characterized by almost entirely Antoni A pattern growth and the absence of Verocay bodies. It was first described in 1981 by Woodruff and co-workers (17), who stressed their benign clinical course and the importance of distinguishing them from malignant peripheral nerve sheath tumors. Subsequent studies have confirmed the benign nature of this tumor (18, 19). As illustrated by the current cases, cellular schwannomas occur most often in the retroperitoneum and display histopathologic features that may result in their misdiagnosis as sarcoma or sarcomatoid carcinoma. These include fascicular architecture, moderate hypercellularity, mitotic figures, and, on rare occasions, small foci of necrosis. Histopathologic features that allow for the correct diagnosis include the presence of ectatic, hyalinized blood vessels; small areas of less cellular Antoni B–type areas; peripheral aggregates of chronic inflammatory cells; and clusters of foamy macrophages. In difficult cases, the finding of intense S-100 protein expression may be useful in confirming this diagnosis.
In all of our cases, the preoperative diagnosis was renal cell carcinoma. The histologic differential diagnosis for renal schwannoma includes angiomyolipoma and leiomyoma, sarcomatoid renal cell carcinoma, malignant peripheral nerve sheath tumor, mesoblastic nephroma of the adult type, and other rare mesenchymal tumors of the kidney. Angiomyolipomas may show great histologic variability, depending on their relative proportions of fat, smooth muscle, and vessels (11). Angiomyolipomas with predominance of the spindled component are most likely to be confused with schwannoma. Histopathologically, however, spindle cell–predominant angiomyolipoma and true leiomyomas are characterized by short fascicles that intersect at right angles and brightly eosinophilic cells with longitudinal striations, blunt-ended nuclei, and perinuclear vacuoles. These are in contrast to the longer fascicles, paler staining cytoplasm, and wavy nuclei of schwannoma. Immunohistochemical demonstration of smooth muscle actin, desmin, and melanosomal proteins such as gp100 (HMB-45) and Melan-A may help to confirm the diagnosis of angiomyolipoma (HMB-45 and actin positive) or leiomyoma (actin and desmin positive). As noted previously, cellular variants of schwannomas may be confused with malignant peripheral nerve sheath tumor. Recognition of the circumscription of cellular schwannomas and the presence of thick-walled blood vessels, foamy histiocytes, and intense S-100 protein expression should allow the distinction of schwannoma from malignant peripheral nerve sheath tumor.
The most important distinction is with sarcomatoid renal cell carcinoma. Whereas renal cell carcinoma may have extensive areas with a spindled growth pattern, careful inspection should reveal focal areas of more typical appearing carcinoma (11). In addition, sarcomatoid renal cell carcinomas are destructive and infiltrative and do not show the well-defined capsule seen in all of our cases of schwannoma. Again, immunohistochemistry may be valuable, as sarcomatoid carcinomas should show at least focal areas of cytokeratin expression and would not be expected to strongly express S-100 protein.
A diagnosis of mesoblastic nephroma (adult type) was considered in Case 2 because of its spindle cell component and the presence of metaplastic bone and cartilage. The immunohistochemical finding of strong S-100 protein positivity, however, argued against that diagnosis, as S-100 is not positive in mesoblastic nephroma (20). Because schwannoma can have prominent paucicellular myxoid areas, myxomas should also be considered in the differential diagnosis. Renal myxoma is an extremely rare benign neoplasm, the existence of which has been questioned in the literature (21, 22). Most of the reported cases have been reinterpreted by Melamed et al. (22) as sarcomas with myxoid features, fibroepithelial polyps, and myxolipoma (22). Recently, three cases of perineurioma of the kidney have been reported (22, 23, 24, 25). Perineuriomas are usually characterized by a storiform proliferation of delicate cells with elongated processes; they would not be expected to show the long fascicles or ectatic blood vessels seen in schwannoma and are epithelial membrane antigen and type IV collagen positive but S-100 protein negative (26).
In conclusion, we present here the largest series to date of renal schwannomas and compare our finding with the few reported in the literature. Renal schwannoma, although rare, must be considered in the differential diagnosis of spindle cell lesions of the kidney. Predilection for the cellular variant to occur in this site may result in a misdiagnosis of the relatively more commonly occurring sarcomatoid renal cell carcinoma. Given the benign clinical course, nephron-sparing surgery would be an option if this diagnosis could be established before radical surgery.
References
Enzinger FM, Weiss SW . Soft tissue tumors, 3rd ed. St. Louis:Mosby; 1995.
Swanson PE, Scheithauer BW, Wick MR . Peripheral nerve sheath neoplasms. Clinicopathologic and immunochemical observations. Pathol Annu 1995;30(Pt 2):1–82.
Phillips CAS, Baumrucker G . Neurilemmoma arising in the left hilus of the kidney. J Urol 1955;73:671–673.
Kuzmina VE . Neurinoma of the kidney capsule. Urologiia 1962;27:52.
Polianichko MF, Zaderin VP, Ratner ON . Nerilemmoma pochki. Urol Nefrol 1982;5:60–62.
Koyama Y, Sasaki M, Deguchi N, Hata M, Tazaki H . Benign schwannoma arising in the kidney. Nishinihon J Urol 1987;49:1843–1846.
Somers WJ, Terpenning B, Lowe FC, Romas NA . Renal parenchymal neurilemoma: a rare and unusual kidney tumor. J Urol 1988;139(1):109–110.
Ma KF, Tse CH, Tsui MS . Neurilemmoma of kidney—a rare occurrence. Histopathology 1990;17(4):378–380.
Kitagawa K, Yamahana T, Hirano S, Kawaguchi S, Mikawa I, Masuda S, et al. MR imaging of neurilemoma arising from the renal hilus. J Comput Assist Tomogr 1990;14(5):830–832.
Ikeda I, Miura T, Kondo I, Kameda Y . Neurilemmoma of the kidney. Br J Urol 1996;78(3):469–470.
Murphy WM, Beckwith JB, Farrow GM, Armed Forces Institute of Pathology (U.S.), Universities Associated for Research and Education in Pathology. Tumors of the kidney, bladder, and related urinary structures. Washington, DC: Armed Forces Institute of Pathology, 1994. Available from the American Registry of Pathology.
Freund ME, Crocker DW, Harrison JH . Neurofibroma arising in a solitary kidney. J Urol 1967;98(3):318–321.
Fein RL, Hamm FC . Malignant schwannoma of the renal pelvis: a review of the literature and a case report. J Urol 1965;94:356–361.
Martinot M, Dupont A, Demaille A . Malignant schwannoma of the kidney. J Urol Med Chir 1960;66:748.
Naslund MJ, Dement S, Marshall FF . Malignant renal schwannoma. Urology 1991;38(5):477–479.
Fabbro MA, Costa L, D'Agostino S, Musi L, Belloli GP . Juxta-adrenal malignant schwannoma. Pediatr Surg Int 1997;12(7):532–534.
Woodruff JM, Godwin TA, Erlandson RA, Susin M, Martini N . Cellular schwannoma: a variety of schwannoma sometimes mistaken for a malignant tumor. Am J Surg Pathol 1981;5(8):733–744.
White W, Shiu MH, Rosenblum MK, Erlandson RA, Woodruff JM . Cellular schwannoma. A clinicopathologic study of 57 patients and 58 tumors. Cancer 1990;66(6):1266–1275.
Fletcher CD, Davies SE, McKee PH . Cellular schwannoma: a distinct pseudosarcomatous entity. Histopathology 1987;11(1):21–35.
Durham JR, Bostwick DG, Farrow GM, Ohorodnik JM . Mesoblastic nephroma of adulthood. Report of three cases. Am J Surg Pathol 1993;17(10):1029–1038.
Kundu AK, Chakraborty AK, Chakraborty S, Das S . Myxoma of the kidney. J Indian Med Assoc 1995;93(12):462.
Melamed J, Reuter VE, Erlandson RA, Rosai J . Renal myxoma. A report of two cases and review of the literature. Am J Surg Pathol 1994;18(2):187–194.
Kahn DG, Duckett T, Bhuta SM . Perineurioma of the kidney. Report of a case with histologic, immunohistochemical, and ultrastructural studies. Arch Pathol Lab Med 1993;117(6):654–657.
Val-Bernal JF, Hernando M, Garijo MF, Villa P . Renal perineurioma in childhood [see comments]. Gen Diagn Pathol 1997;143(1):75–81.
Garcia-Valtuille R, Abascal F, Ortuzar JI, Otero M, Vidal JA . Perineurioma (storiform perineurial fibroma) of the kidney in a child. Eur Radiol 1998;8(5):770–771.
Mentzel T, Dei Tos AP, Fletcher CD . Perineurioma (storiform perineurial fibroma): clinico-pathological analysis of four cases. Histopathology 1994;25(3):261–267.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Alvarado-Cabrero, I., Folpe, A., Srigley, J. et al. Intrarenal Schwannoma: A Report of Four Cases Including Three Cellular Variants. Mod Pathol 13, 851–856 (2000). https://doi.org/10.1038/modpathol.3880150
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880150
Keywords
This article is cited by
-
Giant renal schwannoma with obvious hemorrhage and cystic degeneration: a case report and literature review
BMC Urology (2022)
-
Epithelioid angiosarcoma arising in schwannoma of the kidney: report of the first case and review of the literature
World Journal of Surgical Oncology (2015)
-
Schwannoma of the Kidney
Modern Pathology (2008)
-
Das Schwannom der Niere
Der Urologe (2005)
