
Abstract
Benign peripheral nerve tumors include schwannoma, neurofibroma, and perineurioma, as well as a recently recognized group of tumors with dual patterns of differentiation. The molecular pathogenesis of these so-called “hybrid” tumors remains poorly understood. Following identification of a novel CHD7-VGLL3 fusion gene in a hybrid schwannoma–perineurioma, we evaluated an expanded cohort of this tumor-type—as well as tumors with VGLL3 rearrangement identified from a curated molecular database—to characterize the prevalence of fusion genes among these tumors. Eighteen tumors met the inclusion criteria for this study. RNA sequencing identified VGLL3 rearrangement in 14 of these cases; the partner genes included CHD7 (ten cases), CHD9 (two cases), and MAMLD1 (two cases). Two cases possessed altogether unrelated fusions, including: DST-BRAF and SQSTM1-CDX1 fusion genes. Finally, two cases lacked identifiable fusion products. These findings highlight the molecular diversity of these neoplasms, with frequent rearrangement of VGLL3. More importantly, despite their dual pattern of differentiation, our results reveal the pathogenesis of hybrid schwannoma–perineurioma is unrelated to conventional schwannoma and perineurioma, thereby implying this tumor represents an altogether pathologically distinct entity.
Similar content being viewed by others

Introduction
Benign peripheral nerve tumors are ostensibly divided into three types: schwannoma, neurofibroma, and perineurioma [1]. A subset of neoplasms remain difficult to classify. Indeed, this trichotomy has been challenged with the identification of tumors containing dual patterns of differentiation, so-called “hybrid” tumors (i.e., neurofibroma–schwannoma [2], schwannoma–perineurioma [3, 4], and neurofibroma–perineurioma) [4, 5]. The molecular pathogenesis of peripheral nerve tumors is complex and remains to be fully elucidated. Mutations in NF1 characterize the majority of neurofibromas [6]; schwannomas frequently contain mutations in NF2 [7]; and, intraneural perineuriomas have TRAF7 mutations [8], while their soft tissue counterparts have been reported to harbor mutations in either NF1 or NF2 [9]. The origin of sporadic hybrid peripheral nerve tumors remains unknown.
Following the incidental identification of a novel CHD7-VGLL3 fusion gene in the routine diagnostic evaluation of a hybrid schwannoma–perineurioma, we interrogated a cohort of these tumors to better understand the prevalence, and nature, of fusion genes occurring among these neoplasms.
Materials and methods
Case selection
A CHD7-VGLL3 fusion gene was identified in the index patient in the course of routine diagnostic evaluation. As a result, a retrospective archival review was undertaken at each of the author’s institutions for: (i) tumors classified as hybrid schwannoma–perineurioma, and (ii) tumors containing VGLL3 rearrangement (2017–2020). The original slides were retrieved and rereviewed to confirm the diagnosis based on the established diagnostic criteria [3]. This study was undertaken with institutional research ethics board approval from each of the author’s institutions.
Immunohistochemistry
Formalin-fixed paraffin-embedded tissue sections were stained for S100, SOX10, CD34, neurofilament, epithelial membrane antigen (EMA), claudin-1, GLUT-1, and H3K27me3 using standard techniques, as part of the routine clinical workup at each of the authors’ institutions. Appropriate controls were used throughout. Tumor immunoreactivity was graded semi-quantitatively based on the extent of expression as: diffuse, multifocal, focal, or negative.
RNA sequencing
Formalin-fixed paraffin-embedded tissue sections (either scrolls [3–4 at 10 μm] or tissue scraped from glass slides [4–5 at 4 μm]) were obtained from each case. RNA extraction was performed with the ExpressArt FFPE Clear RNA Ready kit (Amsbio, Cambridge, MA). Libraries were prepared using 20–100 ng total RNA with the TruSight RNA Fusion Panel (Illumina, San Diego, CA), an enrichment-based assay targeting 507 fusion-associated genes. RNA sequencing (RNA-seq) was performed with 76 base-pair paired-end reads on an Illumina MiSeq at 8 samples per flow cell (~3 million reads per sample). The results were analyzed using both the STAR aligner and Manta fusion caller, and the BOWTIE2 aligner and JAFFA fusion caller [10, 11].
Archer™ FusionPlex™ technology was used to develop the MSK-Solid Fusion assay, which is a clinical molecular diagnostic essay performed in a CLIA-accredited laboratory utilizing multiplex polymerase chain reaction to detect oncogenic fusion transcripts involving 62 genes as described previously [12].
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) for CHD7, VGLL3, DST, and BRAF. SQSTM1, and CDX1 was performed as previously outlined in detail [13]. Briefly, bacterial artificial chromosome (BAC) probes were custom designed to flank the target genes, guided by the UCSC genome browser (http://genome.ucsc.edu), and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA; http://bacpac.chori.org) (Supplementary Table 1) [14]. DNA from each BAC was isolated and fluorochrome labeled by nick translation. Formalin-fixed paraffin-embedded tissue (4 μm) were deparaffinized, pretreated, and then hybridized with the denatured probes. After incubating overnight, the slides were rinsed, stained with 4′,6-diamidino-2-phenylindole (DAPI), mounted, and examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany).
Results
Clinical cohort
A total of 19 patients were initially identified; thirteen based on a diagnosis of hybrid schwannoma–perineurioma, and the remainder based on the molecular presence of VGLL3 rearrangement. Following review of the original slides one case with VGLL3 rearrangement was excluded (a low-grade sarcoma with myoid differentiation, and a morphology and immunophenotype incompatible with nerve sheath differentiation [RNA sequencing revealed a TCF12-VGLL3 fusion gene]). Of the final cohort of 18 patients, the average patient age was 36.4 years (range 11–61 years), and 72.2% of tumors occurred in females. 88.9% of lesions were superficial (centered in dermis and/or subcutis) and the average size was 2.3 cm (range 0.7–5.0 cm). The clinical attributes are summarized in Table 1.
Microscopic findings
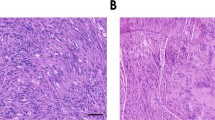
The morphology and immunophenotype were remarkably consistent amongst the cases—including those that were not initially classified as hybrid schwannoma–perineurioma—and consistent with prior reports (Figs. 1–3) [3, 15]. The tumors were composed of an admixture of spindle cells with a storiform-fascicular-whorled architecture with two seemingly distinct cell populations. The predominant cell-type had pale eosinophilic cytoplasm with occasional clear vacuoles and indistinct cell borders; the nuclei were ovoid and plump with occasional pin-point nucleoli. These cells were immunoreactive for S100 and SOX10. The second cell-type had scant eosinophilic cytoplasm with long bipolar processes; the nuclei were slender and elongated, and occasionally undulating. This population of cells appeared to be immunoreactive for CD34, with more variable immunoreactivity for EMA, claudin-1, and GLUT-1 (Table 2). Several cases showed scattered mild nuclear atypia; mitotic activity was inconspicuous (0–2 per 10 HPFs [FD = 0.55 mm]). Entrapped adnexal structures were noted in three cases.

A Scanning magnification showing a circumscribed and unencapsulated neoplasm centered within the subcutaneous adipose tissue. Note entrapped adnexal structures. B, C Intermediate magnifications demonstrating a spindle cells with a storiform-fascicular pattern. The cytoplasm is pale with indistinct borders. D High magnification revealing two distinct nuclear populations. Many cells have plump ovoid nuclei, while a minority are fusiform and elongated. Only mild, likely degenerative, nuclear atypia is present.

Tumors with alternate VGLL3 fusion partners were morphologically and immunophenotypically indistinguishable from those with the more common partner: (A) CHD9-VGLL3 fusion gene, (B) MAMLD1-VGLL3 fusion gene. Similarly, two tumors with altogether different fusion gene products were indistinguishable from those with VGLL3 rearrangement, including cases with a (C) DST-BRAF fusion gene and (D) SQSTM1-CDX1 fusion gene. All images ×400.

A S100, B SOX10, C epithelial membrane antigen and D claudin-1. E Double stain showing alternating parallel layers of cells positive for S100 (red) and epithelial membrane antigen (brown). All images ×200.
Molecular findings
RNA-seq identified VGLL3 rearrangement (exon 2 of 4; NM_016206.4) in 14 cases (77.8%) (Fig. 4). In ten cases this was paired with CHD7 (exon 2 of 38; NM_017780.4); two cases each were partnered with either CHD9 (exon 2 of 39; NM_025134.7), or MAMLD1 (these two tumors contained different breakpoints: exons 3 and 4, of 4; NM_005491). Two cases contained altogether different fusion genes (11.1%): DST (exon 70 of 94; NM_183380.4) with BRAF (exon 11 of 18; NM_004333.6), and SQSTM1 (exon 5 of 8; NM_003900.5) with CDX1 (exon 2 of 3; NM_001804.3). Two cases lacked an identifiable fusion product (11.1%). The morphology and immunophenotype of the DST-BRAF, SQSTM1-CDX1, and fusion-negative cases were indistinguishable from those with VGLL3 rearrangement, thereby precluding separate classification.

A Circos plot demonstrating the chromosomes involved in the various fusion events (image generated using FusionHub) [39]. Exon details for (B) each of the VGLL3 associated fusion partners and (C) unrelated fusion products (mRNA transcripts were extracted and plotted from the R package ggbio) [40]. Note: sky blue arrows and lines indicate the exons involved in the fusion and directions of transcription. D Representative images independently confirming rearrangement of (i) BRAF [three-color FISH break/fusion assay: arrows indicate representative tumor cells with deletion of the telomeric “green” BRAF signal, with corresponding fusion of the centromeric “yellow” BRAF signal to the intragenic “red” DST signal], and (ii) VGLL3 and (iii) CHD7 [break-apart assay: arrows indicate representative tumor cells with break-apart signals].
FISH was done on a subset of cases to independently confirm the novel fusion products (Table 2); testing was also performed on the two negative cases for both CHD7 and VGLL3 which markedly reduced the possibility of a false negative result by RNA-seq for rearrangements involving these two genes. There was no evidence of VGLL3 amplification.
Discussion
Historically a subject of much controversy [16], benign peripheral nerve tumors are now primarily divided into three types—schwannoma, neurofibroma, and perineurioma. A subset of tumors defies conventional classification. Indeed, neoplasms containing permutations of the aforementioned patterns of differentiation, so-called “hybrid” tumors, have recently been recognized by the World Health Organization classification [5]. Following identification of a novel CHD7-VGLL3 fusion gene in a tumor classified as a hybrid schwannoma–perineurioma, we examined a cohort of these tumors by targeted RNA-seq to assess the incidence and nature of fusion drivers amongst this distinctive entity. Our results reveal hybrid schwannoma–perineurioma is generally characterized by recurrent fusion events, with a heterogenous molecular pathogenesis that frequently involves VGLL3 rearrangement.
Most benign peripheral nerve tumors can be accurately classified morphologically. These may be further subdivided into one, or more, histologically distinct subtypes—each also possesses malignant correlates—that can occasionally pose a diagnostic challenge. Perineurioma and schwannoma are neoplasms with predominantly perineurial and Schwann cell differentiation, respectively. Neurofibroma is also considered a neoplasm of Schwann cell origin, although its appearance is attributable to varied contributions from extracellular matrix [17] and several other cell types [18, 19]. Perineural differentiation can be highlighted by immunohistochemical stains including claudin-1, GLUT-1, CD34, and EMA. Schwannian differentiation is identified by staining with S100 and SOX10, and fibroblasts in Antoni “B” regions can occasionally be identified with CD34. Neurofibromas exhibit an admixture of S100 and SOX10, and CD34 staining. Hybrid tumors are composites of two distinct cell populations, which can likewise be recognized by immunohistochemistry. Conceivably this line of reasoning should likewise extend to the molecular level, with hybrid tumors containing established driving mutations in one, or both, cell populations (i.e., NF1 for neurofibroma [6], NF2 for schwannoma [7], and NF1/NF2 for perineurioma [9]). As a matter of fact, in a study of patients with multiple hybrid neurofibromas-schwannomas, there was at least partial monosomy 22—which included the region of NF2—in almost half of patients [20]; this is perhaps unsurprising given the overrepresentation of these tumors amongst patients with neurofibromatosis type 1 and 2, and schwannomatosis [21]. However, to date, the molecular pathogenesis of sporadic hybrid peripheral nerve tumors remains to be elucidated.
Following the discovery of a novel CHD7-VGLL3 fusion gene in a hybrid schwannoma–perineurioma we proceeded to examine a cohort of these tumors by RNA-seq. This revealed VGLL3 rearrangement is common amongst these tumors (77.8%); and this gene has multiple potential partners, including: CHD7 (71.4%), CHD9 (14.3%), and MAMLD1 (14.3%). Other VGLL3 partners presumably exist; in fact, an archive search for tumors with VGLL3 rearrangement identified an unrelated low-grade sarcoma with an in-frame TCF12-VGLL3 fusion gene. Thus, in addition to having multiple potential partners, VGLL3 rearrangement does not appear to be restricted to benign peripheral nerve tumors. Furthermore, novel DST-BRAF and SQSTM1-CDX1 fusion products were identified in two tumors morphologically and immunophenotypically similar to the hybrid schwannoma–perineurioma cohort, suggesting different molecular events may define these tumors. Additional indirect support for this possibility comes from the fact that two cases in our cohort were negative by both RNA-seq and FISH, implying mutation(s) that are not covered by our limited panels. This series did not specifically investigate the possibility of NF1 or NF2 mutations; however, a recent study, using a combination of array comparative genomic hybridization and FISH, did not identify significant overlap between hybrid schwannoma–perineurioma and prior such studies in schwannoma or perineurioma [22].
Vestigial-like family member 3 (VGLL3) is a member of the vestigial-like (VGLL) protein family, serving as a TEA domain–containing transcription factor (TEAD) cofactor [23]. While its physiologic role is uncertain, it appears to promote cell proliferation through activation of the Hippo pathway [23]; interestingly, it has been suggested to have a role in nerve formation and neural crest migration [24]. Amplification has been reported in myxoinflammatory fibroblastic sarcoma [14, 25, 26], along with several other sarcomas [27]; however, to our knowledge, fusion genes involving VGLL3 have not previously been reported. The precise role of members of the chromodomain helicase DNA-binding (CHD) protein family, including CHD7/9, are also unclear [28]. These proteins appear to be involved in transcription regulation, via chromatin remodeling, and rRNA biogenesis [28]. CHD7 promotes neural crest formation [29] and neural progenitor differentiation [30], amongst other diverse associations. Germline mutations in CHD7 are associated with CHARGE syndrome—Coloboma, Heart disease, Atresia choanae, Retarded growth and retarded development and/or CNS anomalies, Genital hypoplasia, and Ear anomalies and/or deafness—which is considered by some a neurocristopathy [29, 31, 32]. CHD9 is expressed by mesenchymal cells and thought to have a role in promoting osteogenic differentiation [33]. A fusion involving this gene was recently reported—a BCOR-CHD9 fusion gene was identified in a renal sarcoma [34]. MAMLD1 is a coactivator in NOTCH signaling [35]. Germline mutations are associated with hypospadias [36], and YAP1-MAMLD1 fusions have been reported in childhood supratentorial ependymomas [37]. Given uncertainty regarding the physiologic roles of these genes, it is difficult to predict the effects of the various fusion genes without functional studies; this is further exacerbated by the potential for seemingly unrelated DST-BRAF and SQSTM1-CDX1 fusion event in these neoplasms. Fusions involving BRAF have been identified in a range of neoplasms (e.g., epithelial, melanocytic, mesenchymal and neural), and these tumors may show a clinical response to RAF or MEK inhibitors; the possible implications of a BRAF fusion in a benign neoplasm are currently uncertain.
While the tumors in our series show dual Schwann cell and perineurial cell differentiation, there is no evidence to suggest a peripheral nerve origin, per se; moreover, the presence of an underlying gene fusion would seem to indicate a distinct pathogenesis. It remains to be established whether one, or both, of the cell types in these tumors contains the fusion product. It is interesting that, in addition to hybrid schwannoma–perineurioma, the initial differential diagnosis of these tumors included variants of neurofibroma, desmoplastic melanoma, and NTRK-rearranged mesenchymal neoplasms (Table 1). While the identification of a fusion may offer diagnostic support in the classification of hybrid schwannoma–perineurioma, it does not exclude the existence of other potential genomic drivers within these neoplasms; moreover, this does not resolve broader conceptual issues related to the ontogenesis of these, and related, neoplasms. Indeed, following a respite from contention, it seems inevitable that next generation sequencing will lead to reinvention in the classification of peripheral nerve tumors. For example, we, and others [38], have identified a case of perineurioma with a GAB1-ABL1 fusion, as well as other molecular events in related tumor-types. Only through the characterization of larger cohorts, with advanced sequencing, expression analysis, and functional studies will be possible to delineate the relationship and molecular breadth these, and related, neoplasms.
In summary, we demonstrate that hybrid schwannoma–perineurioma is frequently characterized by recurrent fusion events, including VGLL3 rearrangement. This finding can be exploited for diagnostic applications when classification is not readily apparent based on morphology and/or immunohistochemistry. More importantly, however, the presence of a discrete molecular event implies these neoplasms—despite evidence of hybrid differentiation—represent a distinct entity with a molecular pathogenesis altogether unrelated to schwannoma and/or perineurioma.
Data availability
The raw RNA sequencing data generated and/or analyzed during the current study are not publicly available due to lack of access to indefinite hosting capabilities. Original data files are available from the corresponding author on reasonable request.
References
Antonescu CR, Scheithauer BW, Woodruff JD. Tumors of the peripheral nervous system. Silver Spring: American Registry of Pathology; 2013.
Feany MB, Anthony DC, Fletcher CD. Nerve sheath tumours with hybrid features of neurofibroma and schwannoma: a conceptual challenge. Histopathology. 1998;32:405–10.
Hornick JL, Bundock EA, Fletcher CD. Hybrid schwannoma/perineurioma: clinicopathologic analysis of 42 distinctive benign nerve sheath tumors. Am J Surg Pathol. 2009;33:1554–61.
Kazakov DV, Pitha J, Sima R, Vanecek T, Shelekhova K, Mukensnabl P, et al. Hybrid peripheral nerve sheath tumors: Schwannoma-perineurioma and neurofibroma-perineurioma. A report of three cases in extradigital locations. Ann Diagn Pathol. 2005;9:16–23.
Stemmer-Rachamimov A, Hornick JL. Hybrid nerve sheath tumour. In: WHO classification of tumours editorial board, editors. Soft tissue and bone tumours. Lyon: International Agency for Research on Cancer; 2020. p. 252–253.
Upadhyaya M, Han S, Consoli C, Majounie E, Horan M, Thomas NS, et al. Characterization of the somatic mutational spectrum of the neurofibromatosis type 1 (NF1) gene in neurofibromatosis patients with benign and malignant tumors. Hum Mutat. 2004;23:134–46.
Agnihotri S, Jalali S, Wilson MR, Danesh A, Li M, Klironomos G, et al. The genomic landscape of schwannoma. Nat Genet. 2016;48:1339–48.
Klein CJ, Wu Y, Jentoft ME, Mer G, Spinner RJ, Dyck PJ, et al. Genomic analysis reveals frequent TRAF7 mutations in intraneural perineuriomas. Ann Neurol. 2017;81:316–21.
Carter JM, Wu Y, Blessing MM, Folpe AL, Thorland EC, Spinner RJ, et al. Recurrent genomic alterations in soft tissue perineuriomas. Am J Surg Pathol. 2018;42:1708–14.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2.
Liu S, Tsai WH, Ding Y, Chen R, Fang Z, Huo Z, et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016;44:e47.
Zhu G, Benayed R, Ho C, Mullaney K, Sukhadia P, Rios K, et al. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod Pathol. 2019;32:609–20.
Kao YC, Sung YS, Zhang L, Chen CL, Vaiyapuri S, Rosenblum MK, et al. EWSR1 fusions With CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol. 2017;41:482–90.
Kao YC, Ranucci V, Zhang L, Sung YS, Athanasian EA, Swanson D, et al. Recurrent BRAF gene rearrangements in myxoinflammatory fibroblastic sarcomas, but not hemosiderotic fibrolipomatous tumors. Am J Surg Pathol. 2017;41:1456–65.
Yang X, Zeng Y, Wang J. Hybrid schwannoma/perineurioma: report of 10 Chinese cases supporting a distinctive entity. Int J Surg Pathol. 2013;21:22–8.
Hajdu SI. Pathology of soft tissue tumors. Lea & Febiger; Philadelphia, PA; 1979.
Fisher ER, Vuzevski VD. Cytogenesis of schwannoma (neurilemoma), neurofibroma, dermatofibroma, and dermatofibrosarcoma as revealed by electron microscopy. Am J Clin Pathol. 1968;49:141–54.
Waggener JD. Ultrastructure of benign peripheral nerve sheath tumors. Cancer. 1966;19:699–709.
Erlandson RA, Woodruff JM. Peripheral nerve sheath tumors: an electron microscopic study of 43 cases. Cancer. 1982;49:273–87.
Stahn V, Nagel I, Fischer-Huchzermeyer S, Oyen F, Schneppenheim R, Gesk S, et al. Molecular analysis of hybrid neurofibroma/schwannoma identifies common monosomy 22 and alpha-T-Catenin/CTNNA3 as a novel candidate tumor suppressor. Am J Pathol. 2016;186:3285–96.
Harder A, Wesemann M, Hagel C, Schittenhelm J, Fischer S, Tatagiba M, et al. Hybrid neurofibroma/schwannoma is overrepresented among schwannomatosis and neurofibromatosis patients. Am J Surg Pathol. 2012;36:702–9.
Hirose T, Kobayashi A, Nobusawa S, Jimbo N. Hybrid schwannoma/perineurioma: morphologic variations and genetic profiles. Appl Immunohistochem Mol Morphol. 2020.
Hori N, Okada K, Takakura Y, Takano H, Yamaguchi N, Yamaguchi N. Vestigial-like family member 3 (VGLL3), a cofactor for TEAD transcription factors, promotes cancer cell proliferation by activating the Hippo pathway. J Biol Chem. 2020;295:8798–807.
Simon E, Theze N, Fedou S, Thiebaud P, Faucheux C. Vestigial-like 3 is a novel Ets1 interacting partner and regulates trigeminal nerve formation and cranial neural crest migration. Biol Open. 2017;6:1528–40.
Hallor KH, Sciot R, Staaf J, Heidenblad M, Rydholm A, Bauer HC, et al. Two genetic pathways, t(1;10) and amplification of 3p11-12, in myxoinflammatory fibroblastic sarcoma, haemosiderotic fibrolipomatous tumour, and morphologically similar lesions. J Pathol. 2009;217:716–27.
Suster D, Michal M, Huang H, Ronen S, Springborn S, Debiec-Rychter M, et al. Myxoinflammatory fibroblastic sarcoma: an immunohistochemical and molecular genetic study of 73 cases. Mod Pathol. 2020;33:2520–33.
Helias-Rodzewicz Z, Perot G, Chibon F, Ferreira C, Lagarde P, Terrier P, et al. YAP1 and VGLL3, encoding two cofactors of TEAD transcription factors, are amplified and overexpressed in a subset of soft tissue sarcomas. Genes Chromosomes Cancer. 2010;49:1161–71.
Zentner GE, Hurd EA, Schnetz MP, Handoko L, Wang C, Wang Z, et al. CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Hum Mol Genet. 2010;19:3491–501.
Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463:958–62.
Yao H, Hannum DF, Zhai Y, Hill SF, Albanus RD, Lou W, et al. CHD7 promotes neural progenitor differentiation in embryonic stem cells via altered chromatin accessibility and nascent gene expression. Sci Rep. 2020;10:17445.
Pagon RA, Graham JM Jr., Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99:223–7.
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. 214800, 2020. https://omim.org/.
Shur I, Benayahu D. Characterization and functional analysis of CReMM, a novel chromodomain helicase DNA-binding protein. J Mol Biol. 2005;352:646–55.
Kao YC, Sung YS, Argani P, Swanson D, Alaggio R, Tap W, et al. NTRK3 overexpression in undifferentiated sarcomas with YWHAE and BCOR genetic alterations. Mod Pathol. 2020;33:1341–9.
Lin SE, Oyama T, Nagase T, Harigaya K, Kitagawa M. Identification of new human mastermind proteins defines a family that consists of positive regulators for notch signaling. J Biol Chem. 2002;277:50612–20.
Chen Y, Thai HT, Lundin J, Lagerstedt-Robinson K, Zhao S, Markljung E, et al. Mutational study of the MAMLD1-gene in hypospadias. Eur J Med Genet. 2010;53:122–6.
Andreiuolo F, Varlet P, Tauziede-Espariat A, Junger ST, Dorner E, Dreschmann V, et al. Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol. 2019;29:205–16.
Panagopoulos I, Gorunova L, Andersen K, Tafjord S, Lund-Iversen M, Lobmaier I, et al. Recurrent fusion of the GRB2 associated binding protein 1 (GAB1) gene with ABL proto-oncogene 1 (ABL1) in benign pediatric soft tissue tumors. Cancer Genom Proteom. 2020;17:499–508.
Panigrahi P, Jere A, Anamika K. FusionHub: a unified web platform for annotation and visualization of gene fusion events in human cancer. PLoS ONE. 2018;13:e0196588.
Yin T, Cook D, Lawrence M. ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol. 2012;13:R77.
Funding
Panov 2 Research Fund (BCD); P50 CA 140146-01 (CRA), P50 CA217694 (CRA), P30 CA008748, Cycle for Survival (CRA).
Author information
Authors and Affiliations
Contributions
BCD and JLH performed study concept and design; BCD performed writing and revision of the paper; BCD, JLH, CRA, DS, LZ, and NDA provided acquisition, analysis and/or interpretation of data; CDMF, EGD, and IL provided material support; and, LZ and DS provided technical support. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Conflict of interest
BCD has, in the past, received a limited number of RNA-seq test kits pro bono from Illumina. These kits were not applied to this work. The other authors declare no competing interests.
Ethical approval
This study was performed following institutional Research Ethics Board Approval (Mount Sinai Hospital, 17-0103-E). This study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Dickson, B.C., Antonescu, C.R., Demicco, E.G. et al. Hybrid schwannoma–perineurioma frequently harbors VGLL3 rearrangement. Mod Pathol 34, 1116–1124 (2021). https://doi.org/10.1038/s41379-021-00783-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00783-0
This article is cited by
-
What is new in intraneural perineurioma?
Acta Neurochirurgica (2023)
-
RNA-sequencing of myxoinflammatory fibroblastic sarcomas reveals a novel SND1::BRAF fusion and 3 different molecular aberrations with the potential to upregulate the TEAD1 gene including SEC23IP::VGLL3 and TEAD1::MRTFB gene fusions
Virchows Archiv (2022)
-
Hybrid tumors with perineurioma components: a systematic review of the literature and illustrative case
Acta Neurochirurgica (2022)
