
Abstract
Neoplasms with mixed carcinomatous and sarcomatous growth patterns occur in many organs and tissues. The pathogenesis of these cancers is thought to be either the result of two independent neoplastic processes merging to form a single tumor, or a neoplasm of monoclonal origin that develops phenotypic diversity. To address this issue, we characterized molecular alterations in separately microdissected epithelial and sarcomatous areas in three cases of pancreatic mucinous cystic neoplasms with sarcomatous stroma. Using microsatellite markers for six chromosomal loci commonly deleted in infiltrating ductal adenocarcinomas of the pancreas, we found genetic alterations to be virtually identical between the sarcomatous and epithelial components of two of the three neoplasms. In the third neoplasm, we found allelic losses and retentions to be identical at five of the six chromosomal loci, but at a single locus, we noted allelic loss in the neoplastic epithelial component but not the sarcomatous component. The same neoplasms were also analyzed for activating point mutations in codon 12 of the K-ras gene by using mutant-enriched polymerase chain reaction and allele-specific oligonucleotide hybridization. A K-ras mutation was identified in the epithelial component of one of the three neoplasms (the same tumor with an additional allelic loss in the neoplastic epithelial cells), but the sarcomatous component of this tumor was wild-type at codon 12 of K-ras, as were both components of the other two neoplasms. Overall, these results suggest a monoclonal origin with subsequent divergence of the neoplastic epithelial and sarcomatous portions of these neoplasms.
Similar content being viewed by others
INTRODUCTION
Neoplasms with mixed epithelial and sarcomatous differentiation are relatively common and have been reported in a variety of organs including the breast, lung, uterus, bladder, skin, head and neck, pancreas, and prostate (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12). In some of these neoplasms, a transition between the epithelial and stromal components can be readily recognized, whereas in others, the carcinomatous and sarcomatous portions of the tumor are distinct.
The pathogenesis of these neoplasms, particularly those with distinct epithelial and mesenchymal components, has remained a subject of controversy (1). One possible explanation for the mixed growth pattern is that two neoplasms of independent pathogenesis converge (collision tumor), whereas the more commonly held hypothesis is that a single neoplasm of monoclonal origin develops divergent patterns of gene expression during the evolution of the neoplasm (1).
Modern molecular genetic techniques combined with careful microdissection now make it possible to resolve this controversy. If the two components are independent processes, then they should show distinct genetic alterations. If, conversely, they represent a single neoplasm of monoclonal origin that only later develops divergent differentiation, then one would expect the two components to show shared genetic alterations.
The pancreatic mucinous cystic neoplasm with sarcomatous stroma is an ideal tumor type to study such divergent differentiation for two reasons. First, these neoplasms have been well characterized microscopically and immunohistochemically (13). Second, the epithelial component lines easily identified cystic spaces, greatly increasing the ease and precision of microdissection. The two components of the neoplasm can therefore be separately analyzed.
We conducted a molecular analysis of three pancreatic mucinous cystic neoplasms with sarcomatous stroma. Our analysis included assessment of chromosomal alleles at six loci commonly affected by loss of heterozygosity in infiltrating ductal carcinomas of the pancreas and mutational analysis of codon 12 of the K-ras gene, which is frequently mutated in ductal pancreas cancer (14, 15, 16). By selectively microdissecting sarcomatous, neoplastic epithelial, and normal cells from the tissues, we determined whether phenotypically different areas of the neoplasm shared a molecular genetic pathogenesis.
MATERIALS AND METHODS
Tissues
Formalin-fixed paraffin-embedded blocks were obtained from three well-characterized and previously reported pancreatic mucinous cystic neoplasms with sarcomatous stroma (13). The neoplastic epithelial and sarcomatous components of these neoplasms were separately microdissected, as has been described (17, 18, 19). Briefly, the blocks were sectioned at 12 μ, stained with hematoxylin and eosin, visualized with an inverted microscope, and dissected using a 26-gauge needle. First, subepithelial stroma was removed, and then inflammatory cells and other non-neoplastic tissues were removed, allowing for clean dissection of sarcomatous and neoplastic epithelial cells. Each component was microdissected to more than 95% cellularity. Normal tissue from each case was similarly microdissected from adjacent non-neoplastic stroma, acini, or inflammatory cells. The microdissected tissues were then digested overnight in buffer containing 0.5% NP40 and 200 μg/mL proteinase K. This lysate was then used directly in polymerase chain reactions (PCR).
Microsatellite Analysis
Polymorphic microsatellite markers were selected to amplify the following chromosomal regions with known tumor suppressor genes or frequent loss of heterozygosity in infiltrating ductal cancers of the pancreas: D9S156 at 9p21 (the p16 gene); TP53 and CHRNB1 at 17p13 (the p53 gene); D18S474, D18S46, and D18S4655 (the DPC4 gene); D3S1286 and D3S1293 (chromosome 3); D6S292 and D6S261(chromosome 6); and D8S264 and D8S261. All microsatellite marker primers were purchased from Research Genetics (Huntsville, AL) and amplified using 30 to 35 cycles of PCR and annealing temperatures of 56 to 58° C. PCR products were then separated by denaturing gel electrophoresis and visualized by autoradiography. Loss of heterozygosity was scored when one allele showed a greater than 80% reduction in intensity in tumor DNA compared with normal DNA. All findings were validated by a repeat reaction with the same primers or a similar result in a reaction with an adjacent primer.
K-ras Gene Mutations
The microdissected foci were evaluated for activating point mutations in codon 12 of K-ras as has been previously described (20). Briefly, DNA was isolated and subjected to PCR amplification using primers A (5′-ACT GAA TAT AAA CTT GTG GTA GTT GGA CCT-3′) and D (5′-TCA TGA AAA TGG TCA GAG AAA CC-3′). This set of primers introduces an Mva 1 restriction site in PCR products derived from wild-type codon 12 alleles. Mutant enrichment was performed by digestion of the PCR product using the restriction enzyme Mva 1. This enzyme cleaves the wild-type but not the mutant alleles of K-ras. A 1-μl aliquot of the digested first PCR was then subjected to a second round of amplification using the semi-nested primers A and B (5′-TCA AAG AAT GGT CCT GGA CC-3′). The resulting DNA fragments were then spot-blotted onto seven different nylon membranes, and each of these separate membranes was hybridized with an allele-specific oligonucleotide probe for the wild-type K-ras sequence or for one of the six possible mutations in codon 12 of K-ras. Positive controls included cloned wild-type and mutant sequences, and no DNA was added in the negative controls. All allele-specific oligonucleotide analyses were performed in duplicate.
RESULTS
Pathology and Clinical Features of Tumors
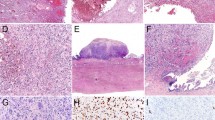
The clinical, radiographic, gross, microscopic, and immunohistochemical staining characteristics of the three cases have been previously reported (13). Briefly, all three neoplasms occurred in the tail of the pancreas, all three were large and multicystic, and all three contained a mucin producing epithelial component (Figs. 1A, B) and a sarcomatous component (Figs. 1C, D) (13). The epithelial component was composed of columnar to cuboidal cells with varying degrees of atypia ranging from benign to atypical to overtly malignant (13). The epithelial component in all three cases expressed cytokeratin (Fig. 1B), epithelial membrane antigen, and carcinoembryonic antigen (13). The sarcomatous components in all three cases variably expressed vimentin and actin, and in two cases it expressed estrogen and progesterone receptors (13). One patient had omental metastases composed entirely of sarcomatous stroma.

The epithelial (A and B) and sarcomatous (C and D) components of a mucinous cystic neoplasm with sarcomatous stroma. The epithelial component is composed of tall columnar mucin producing cells (A) that express cytokeratin (B). By contrast, the sarcomatous component was composed of spindle shaped cells with pleomorphic nucleic and a high mitotic rate (C and D). (A, C, and D, hematoxylin and eosin, 200 ×; B, immunohistochemical stain for cytokeratin, 200 ×).
Loss of Heterozygosity
All three neoplasms were found to have a polymorphism for at least one microsatellite marker at each of the six chromosomal loci studied, and all three were therefore informative at all six loci. All three cases showed retention of chromosomal arm 3p and loss of heterozygosity (LOH) of chromosomal arm 6q. In addition, LOH was identified in one of three cases for chromosomal arm 8p, in two of three cases for chromosomal arm 9p, in two of three cases for chromosomal arm 17p, and in one of three cases for chromosomal arm 18q (Table 1, Fig. 2).

Loss of heterozygosity in mucinous cystic neoplasms with sarcomatous stroma. Representative microsatellite amplifications are shown for each of the three cases. N, normal cell DNA; S, sarcomatous stroma DNA; E, neoplastic epithelial cell DNA. Dots to the left of each panel designate positions of alleles, and arrows to right of panels designate alleles lost in tumor samples.
Remarkably, with one exception, the patterns of allelic loss, including both the chromosome arms lost and the specific alleles lost, were identical for the sarcomatous and neoplastic epithelial components of these neoplasms. The single exception to this observation was the LOH of chromosomal arm 9p seen in the neoplastic epithelial cells, but not the stromal cells, of Case 3.
Mutational Analysis of the K-ras Oncogene
Samples of the neoplastic epithelial and sarcomatous components from each neoplasm were individually evaluated for activating point mutations in codon 12 of the K-ras gene. For Cases 1 and 2, no K-ras mutations were seen in either component of the tumor; however, the stromal component was not amplifiable in Case 1. In Case 3, a GGT → GAT mutation of codon 12 was seen in the neoplastic epithelial cells, but not in the stromal cells. These results parallel the finding of LOH at 9p in the neoplastic epithelial cells, but not the sarcomatous cells, of Case 3 (Table 1, Fig. 3).

Representative autoradiograph of the K-ras codon 12 point mutation analysis. Four nylon membranes, each hybridized with a different radioactive-labeled oligonucleotide specific for the sequence of the wild-type codon 12 (left lane), and six possible mutations, three of them depicted. On each membrane in the left lane, the nonenriched polymerase chain reaction products are represented and in the right lane the mutant-enriched PCR products. co, hybridization controls, on each membrane cloned DNA fragments with a known codon 12 sequence complementary to the labeled oligonucleotides are used for the hybridization of that membrane; 1 and 2, DNA isolated from the epithelial and stromal component respectively of Case 2, both with a wild-type K-ras codon 12 sequence; 3 and 4, DNA isolated from the epithelial (3) and stromal (4) component of Case 3 with a mutation in the epithelial component (GGT to GAT), resulting in an amino acid change from glycine to aspartic acid pla, placenta DNA, control for nonspecific hybridization; H2O, negative water control; 1:00 and 1:1000, positive controls containing DNA from cell suspensions with mutant wild-type ratios of 1:100 and 1:1000; the suspensions were made of the human colon cancer cell line SW 480 with a homozygous GGT to GTT mutation at codon 12 of K-ras (glycine to valine) and the human colon cancer cell line HT 29 with wild-type K-ras.
DISCUSSION
The pathogenesis of human cancers is thought to be the result of genetic instability that produces multiple genetic alterations, of which some activate growth-promoting genes (oncogenes) and others inactivate growth-suppressing genes (tumor suppressor genes) (21, 22). These genetic alterations most likely occur in a series of steps, with clonal expansion of cells occurring when genetic alteration results in a selective growth advantage (23). In most cancers, neoplastic cells can be demonstrated to be clonal in origin by virtue of all cells having a “fingerprint” of identical genetic changes. However, genetic heterogeneity also occurs even though the cancers, on the basis of sharing a majority of genetic changes, appear to be clonal in origin (24). For example, substantial genetic heterogeneity has been demonstrated in colorectal neoplasms, intraductal cancers of the breast, and in intraductal papillary-mucinous neoplasms of the pancreas (17, 18, 19, 24).
The purpose of this study was to characterize the molecular changes in pancreatic mucinous cystic neoplasms with sarcomatous stroma, specifically to determine whether or not the neoplastic epithelial and sarcomatous portions of the neoplasm have genetic alterations consistent with a common clonal origin. These neoplasms were selected for study because they are otherwise well characterized and because the neoplastic epithelial and sarcomatoid cells are clearly distinct from one another and, thus, can be individually sampled by microdissection.
In two of the neoplasms examined, the patterns of allelic losses and retentions were identical and both components were found to be wild-type for K-ras. These results are consistent with the two cell types sharing a common clonal origin. In the third neoplasm, losses and retentions were the same for the two components at five of the six chromosomal loci, and at one locus (9p21), LOH was seen in the neoplastic epithelial cells, but not the sarcomatous cells. Genetic heterogeneity was also seen in this tumor for K-ras mutations, which were present in the neoplastic epithelial cells, but not the sarcomatous component. These results are consistent with the neoplastic epithelial and sarcomatous components sharing a common clonal origin, with superimposed genetic heterogeneity that occurred during tumor progression.
It is somewhat surprising that more genetic heterogeneity was not seen, given the phenotypic diversity of these cancers. It is likely that the phenotypic differences in the two regions of the cancer are the result of genetic alterations at loci and genes that we did not investigate.
The allelotype at six chromosomal loci and mutational analysis of K-ras in these cases also allowed us to compare the genetic alterations of mucinous cystic neoplasms with sarcomatous stroma of the pancreas with the more common infiltrating ductal carcinomas of the pancreas (14, 15, 16). The genetic alterations of ductal carcinomas have been well characterized, and the majority of these cancers have activating point mutations of codon 12 of K-ras, inactivation of p16 (on chromosomal arm 9p), mutations of p53 (on chromosomal arm 17p), and mutations of DPC4 (on chromosomal arm 18q) (16, 25, 26, 27, 28, 29, 30, 31). Although our sample size was small, only one of these mucinous neoplasms harbored a K-ras mutation compared with a frequency of K-ras mutations in ductal pancreatic cancers of 90% (16). Similarly, other genetic alterations, including LOH of 18q, were not as prevalent in the pancreatic mucinous cystic neoplasms with sarcomatous stroma as they are in infiltrating ductal cancers of the pancreas. These molecular differences correlate with substantial differences in the pathologic and clinical features of the two neoplasms.
Although Thompson et al. (32) did not identify any K-ras mutations in over 50 mucinous cystic neoplasms of the pancreas that they analyzed, we identified an activating point mutation in codon 12 of K-ras in one of three mucinous cystic neoplasms with sarcomatous stroma. This difference may reflect differences in methodologies or differences in the biology of these closely related neoplasms (33).
In conclusion, neoplasms with both epithelial and sarcomatous differentiation are rare, but they provide a unique opportunity to study divergent differentiation in a neoplasm. Although mucinous cystic neoplasms with sarcomatous stroma appear to have patterns of genetic alterations distinct from the more common ductal cancers of the pancreas, genetic alterations are largely shared between the neoplastic epithelial and sarcomatous cells, suggesting that these two components of the tumors share a common clonal origin.
References
Wick MR, Swanson PE . Carcinosarcomas: current perspectives and an historical review of nosological concepts. Semin Diagn Pathol 1993; 10: 118–127.
Zarbo RJ, Crissman JD, Venkat H, Weiss MA . Spindle-cell carcinoma of the upper aerodigestive tract mucosa. An immunohistologic and ultrastructural study of 18 biphasic tumors and comparison with seven monophasic spindle-cell tumors. Am J Surg Pathol 1986; 10: 741–753.
Leman BI, Walker PD . Sarcomatoid carcinoma of the pancreas. Am J Clin Pathol 1981; 76: 351.
Bauer TW, Rostock RA, Eggleston JC, Baral E . Spindle cell carcinoma of the breast: four cases and review of the literature. Hum Pathol 1984; 15: 147–152.
Humphrey PA, Scroggs MW, Roggli VL, Shelburne JD . Pulmonary carcinomas with a sarcomatoid element: an immunocytochemical and ultrastructural analysis. Hum Pathol 1988; 19: 155–165.
Young RH, Wick MR, Mills SE . Sarcomatoid carcinoma of the urinary bladder: a clinicopathologic analysis of 12 cases and review of the literature. Am J Clin Pathol 1988; 90: 653–661.
Ro JY, Chen JL, Lee JS, Sahin AA, Ordonez NG, Ayala AG . Sarcomatoid carcinoma of the lung. Immunohistochemical and ultrastructural studies of 14 cases. Cancer 1992; 69: 376–386.
Bitterman P, Chun B, Kurman RJ . The significance of epithelial differentiation in mixed mesodermal tumors of the uterus. A clinicopathologic and immunohistochemical study. Am J Surg Pathol 1990; 14: 317–328.
Clement PB, Scully RE . Uterine tumors with mixed epithelial and mesenchymal elements. Semin Diagn Pathol 1988; 5: 199–222.
Reuter VE . Sarcomatoid lesions of the urogenital tract. Semin Diagn Pathol 1993; 10: 188–201.
Lauwers GY, Schevchuk M, Armenakas N, Reuter VE . Carcinosarcoma of the prostate. Am J Surg Pathol 1993; 17: 342–349.
Westra WH, Sturm PJ, Drillenburg P, Choti MA, Klimstra DS, Abores-Saavedra J, et al. K-ras oncogene mutations in osteoclast-like giant-cell tumors of the pancreas and liver: genetic evidence to support origin from the duct epithelium. Am J Surg Pathol 1998; 22: 1247–1254.
Wenig BM, Albores-Saavedra J, Buetow PC, Heffess CS . Pancreatic mucinous cystic neoplasm with sarcomatous stroma: a report of three cases. Am J Surg Pathol 1997; 21: 70–80.
Hahn SA, Seymour AB, Hoque ATMS, Schutte M, da Costa LT, Redston MS, et al. Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res 1995; 55: 4670–4675.
Seymour A, Hruban RH, Redston MS, Caldas C, Powell SM, Kinzler KW, et al. Allelotype of pancreatic adenocarcinoma. Cancer Res 1994; 54: 2761–2764.
Hruban RH, van Mansfeld ADM, Offerhaus GJA, van Weering DHJ, Allison DC, Goodman SN, et al. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol 1993; 143: 545–554.
Fujii H, Inagaki M, Kasai S, Miyokawa N, Tokusashi Y, Gabrielson E, et al. Genetic progression and heterogeneity in intraductal papillary-mucinous neoplasms of the pancreas. Am J Pathol 1997; 151: 1447–1454.
Fujii H, Marsh C, Cairns P, Sidransky D, Gabrielson E . Genetic divergence in the clonal evolution of breast cancer. Cancer Res 1996; 56: 1493–1497.
Fujii H, Szumel R, Marsh C, Zhou W, Gabrielson E . Genetic progression, histological grade and allelic loss in ductal carcinoma in situ of the breast. Cancer Res 1996; 56: 5260–5265.
Hruban RH, Sturm PDJ, Slebos RJC, Wilentz RE, Musler AR, van Velthuysen ML, et al. Can K-ras codon 12 mutations be used to distinguish benign bile duct proliferations from metastases in the liver? A molecular analysis of 101 liver lesions from 93 patients. Am J Pathol 1997; 151: 943–949.
Fearon ER, Vogelstein B, Kinzler KW, editors. The genetic basis of human cancer. New York: McGraw Hill; 1997. pp. 229–236.
Kinzler KW, Vogelstein B . Gatekeepers and caretakers. Nature 1997; 386: 761–763.
Fearon ER, Vogelstein B . A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–767.
Tsao J, Tavare S, Salovaara R, Jass JR, Aaltonen LA, Shibata D . Colorectal adenoma and cancer divergence. Am J Pathol 1999; 154: 1815–1824.
Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994; 54: 3025–3033.
DiGiuseppe JA, Hruban RH, Goodman SN, Polak M, van den Berg FM, Allison DC, et al. Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994; 101: 684–688.
Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994; 8: 27–32.
Hahn SA, Schutte M, Hoque ATMS, Moskaluk CA, daCosta LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996; 271: 350–353.
Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res 1997; 57: 1731–1734.
Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997; 57: 3126–3130.
Hruban RH, Petersen GM, Ha PK, Kern SE . Genetics of pancreatic cancer: from genes to families. Surg Oncol Clin N Am 1998; 7: 1–23.
Thompson LDR, Becker RC, Przygodzki RM, Adair CF, Heffess CS . Mucinous cystic neoplasm (mucinous cystadenocarcinoma of low-grade malignant potential) of the pancreas: a clinicopathologic study of the pancreas. Am J Surg Pathol 1999; 23: 1–16.
Wilentz RE, Albores-Saavedra J, Zahurak M, Talamini MA, Yeo CJ, Cameron JL, et al. Pathologic examination accurately predicts prognosis in mucinous cystic neoplasms of the pancreas. Am J Surg Pathol 1999; 23: 1320–1327.
Acknowledgements
Supported in part by National Institutes of Health Grant P50CA-62924 and NOW Grant 950–10-625.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
van den Berg, W., Tascilar, M., Offerhaus, G. et al. Pancreatic Mucinous Cystic Neoplasms with Sarcomatous Stroma: Molecular Evidence for Monoclonal Origin with Subsequent Divergence of the Epithelial and Sarcomatous Components. Mod Pathol 13, 86–91 (2000). https://doi.org/10.1038/modpathol.3880013
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880013
Keywords
This article is cited by
-
Ovarian mucinous cystic tumor of borderline malignancy with a mural nodule of anaplastic spindle cell carcinoma: a case report
Journal of Ovarian Research (2013)
-
Pancreatic mucinous cystic neoplasm with sarcomatous stroma metastasizing to liver: a case report and review of literature
World Journal of Surgical Oncology (2013)
-
Long-term recurrence-free survival in a patient with pancreatic carcinosarcoma: A case report with a literature review
Medical Oncology (2012)
-
Neue Einblicke in die Entstehung des Pankreaskarzinoms
Der Pathologe (2012)
-
Solitary splenic metastasis from ovarian carcinosarcoma: a case report
Journal of Medical Case Reports (2011)
