
Abstract
A preferential use of one particular immunoglobulin variable heavy chain gene, VH3–21, has recently been reported in mantle cell lymphoma, where almost all of these VH3–21+ mantle cell lymphomas showed usage of the same light chain Vλ gene (Vλ3–19) and also had a tendency towards improved prognosis. These findings suggested that VH3–21+ mantle cell lymphomas constitute a distinct subgroup, possibly with antigen stimulation involved in disease pathogenesis. In this study, we applied the comparative genomic hybridization (CGH) method on 37 mantle cell lymphoma tumors in order to investigate if the VH3–21+ tumors are different at the genomic level. Interestingly, VH3–21+ mantle cell lymphomas (n=14) showed significantly fewer genomic aberrations (mean 2.4) compared to non-VH3–21 mantle cell lymphomas (n=23) (mean 4.9). The chromosomal aberrations identified in our study were generally in accordance with previous CGH studies of mantle cell lymphoma; the most frequent aberration was complete or partial loss of chromosome 13, followed by recurrent losses within 6q, 9p, 9q and 11q and frequent gains in 3q, 7p, 8q and 15q. Deletions within 8p and 9p as well as gains in 7p and 15q were found exclusively in the non-VH3–21-utilizing tumors. In summary, VH3–21+ mantle cell lymphomas demonstrated both a lower number and a different spectrum of genomic aberrations than mantle cell lymphoma in general, thus supporting the hypothesis that VH3–21+ mantle cell lymphomas constitute a new subgroup. The findings presented in this report may explain the tendency for a better clinical outcome for patients whose tumors utilize VH3–21.
Similar content being viewed by others

Main
Mantle cell lymphoma is an aggressive lymphoma with a median survival of 3–5 years. Despite the cytological appearance of a small-cell population that previously was misjudged as a sign of low-grade malignancy, mantle cell lymphoma is now considered to have the worst prognosis of all lymphoma types. For the majority of cases, naïve pregerminal center B cells, located in primary lymphoid follicles or the mantle zone of secondary follicles are considered to give rise to the transformed cells.1, 2 Cytogenetically, mantle cell lymphoma is characterized by a reciprocal translocation, t(11;14)(q13;q32), which is found in virtually all cases.3 Through this rearrangement, CCND1/PRAD-1/BCL1 on chromosome 11 is juxtaposed to the active IGH enhancer on chromosome 14. As a consequence, the cyclin D1 protein is overexpressed (a cell cycle regulator normally not expressed in B cells) causing a disturbance in the control of the transition from G1 to S phase.4 Recently, a small subset of mantle cell lymphoma cases that do not have this genetic abnormality have been described, which instead express either cyclin D2 or D3, underlining the importance of the dysregulation of cell cycle control at this check point.5 However, the same study shows that these groups do not differ in survival time. Mantle cell lymphoma can be broadly categorized into one of two morphological types; classical mantle cell lymphoma with small- to medium-sized cells or a blastoid variant with larger cells that is associated with high mitotic index and even more aggressive behavior.3
Characterization of variable heavy chain (VH) gene usage has recently been performed in mantle cell lymphoma, revealing that two particular VH gene segments of the immunoglobulin (Ig) locus were preferentially used, namely VH3–21 (18%) and VH4–34 (16%).2, 6 Interestingly, the tumor samples with VH3–21 gene usage almost exclusively expressed lambda light chains, and also in most cases utilized the same Vλ light chain gene (Vλ3–19).6 In addition, this group has been shown to have a significantly better prognosis in one report and in other studies displayed a clear tendency towards improved survival compared to mantle cell lymphomas utilizing other VH genes.6, 7, 8 Thus, considering that one-fifth of mantle cell lymphoma use the same VH gene (VH3–21), display common molecular characteristics, and have better clinical outcome, it is likely that these patients constitute a subgroup of mantle cell lymphoma, where antigen(s) could possibly play a role in the pathogenesis of the disease. However, since the VH genes are unmutated in the VH3–21+ subset and thus show no signs of transit through the germinal center, the hypothesized antigen stimulation most likely gives rise to a T-cell independent reaction.
Using the comparative genomic hybridization (CGH) technique, it is possible to screen entire tumor genomes for losses or gains of chromosomal regions without requiring cell culturing and metaphase preparation from the tumor itself. Earlier CGH studies indicate that mantle cell lymphoma constitutes a genetically homogeneous subgroup of lymphoma characterized by recurrent aberrations, in particular gains within 3q and 8q and losses in 6q, 9p, 11q and 13q.9, 10, 11, 12 An increased number of aberrations in addition to complex karyotypes have also been reported to be associated with poor prognosis in mantle cell lymphoma.10 In the current series, we have applied the CGH technique to investigate whether the genomic architecture differs between the VH3–21+ group as compared to mantle cell lymphoma in general.
Materials and methods
Patients and Tumor Samples
The clinical data for the patients included are shown in Tables 1 and 2. In all, 37 tumor samples from mantle cell lymphoma patients diagnosed between 1988 and 2001 were identified from the registries of the Departments of Pathology at Uppsala University Hospital (n=15), Lund University Hospital (n=10), Karolinska Hospital (n=8), Umeå University Hospital (n=2), and Huddinge University Hospital (n=2), Sweden. The samples, all taken at diagnosis except case 21, were selected from a larger cohort included in a previous study.6 Mantle cell lymphoma diagnosis was based on morphologic and immunophenotypic criteria according to the World Health Organization (WHO) classification.3 Cyclin D1 overexpression was confirmed by immunohistochemistry in 33 cases using a cyclin D1 antibody, clone P2D11F11 (Novocastra, Newcastle upon Tyne, UK). Paraffin sections were subjected to antigen retrieval with Tris–EDTA pH 9 in a microwave oven for 10 min on full power, then 15 min at 350 W. Slides were stained using an automated immunostainer (Autostainer Plus, DAKO, Carpinteria, CA, USA) with a dilution factor of 1:20 used for the antibody. In the remaining four cases, the t(11;14)(q13;q32) was demonstrated by interphase FISH analyses according to the manufacturer's recommendations using the Vysis LSI IGH/CCND1 probe set for t(11;14) (Vysis Inc, Downers Grove, IL, USA). All mantle cell lymphoma patients were diagnosed at the individual departments and were also centrally reviewed by an experienced hematopathologist.6 Survival information was obtained from medical records and local Swedish cancer registries.
VH Gene Analysis
All samples have previously been analyzed for VH gene rearrangements and a selection of 14 VH3–21+ cases and 23 non-VH3–21 cases was made from a cohort of 110 mantle cell lymphomas.6 In brief, clonal PCR products were sequenced directly in 28 cases, whereas subcloning was performed in nine cases as described earlier.6 The consensus primers used for VH gene family-specific PCR and the PCR conditions have been described elsewhere.13 The sequences were aligned with published IGH sequences retrieved from the GenBank, V-BASE and IMGT databases. The VH gene sequence was considered mutated if the deviation from the corresponding germline gene was greater than 2%.
CGH
High-molecular-weight DNA was prepared from fresh frozen tumor tissues using a standard protocol for phenol–chloroform extraction as previously outlined2 or with the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA), according to the recommendations from the manufacturer.
CGH was performed as previously described14 with minor modifications. Briefly, tumor DNA samples were labeled with fluorescein isothiocyanate (FITC)-12-dUTP (DuPont NEN, Boston, MA, USA), and normal reference DNA was labeled with Spectrum Red (Vysis, Inc.), by nick translation. The nick translation conditions were adjusted to obtain DNA fragments of 300–2000 base pairs in size. Tumor and reference DNA were mixed together with unlabeled Cot-1-DNA (Invitrogen, Groningen, the Netherlands), denatured and applied onto slides with denatured metaphases of normal lymphocytes (Vysis, Inc.). After hybridization at 37°C for 48 h, the slides were first washed in 0.4 × SSC (standard saline citrate)/0.3% Nonidet P-40 at 74±1°C for 2 min, then in 2 × SSC/0.1% Nonidet P-40 at room temperature for 2 min, and finally in sterile water for 2 min. After air drying, the slides were counterstained with 4,6-diamidino-2-phenylindole (DAPI) (Vysis, Inc.) in an antifade solution (Vectashield, Vector Inc., Burlingame, CA, USA).
Digital Image Analysis
A minimum of 10 three-color digital images (DAPI, FITC and Spectrum Red fluorescence) were captured from each hybridization using an Axioplan 2 epifluorescence microscope (Carl Zeiss Jena GmbH, Jena, Germany) equipped with a Sensys cooled charge-coupled device (CCD) camera (Photometrics), and images were analyzed with the isis/CGH software (Metasystems, Altlussheim, Germany). Relative DNA sequence copy number changes were detected by analyzing the hybridization intensities of tumor and normal DNA along the length of all chromosomes in each metaphase spread. The absolute fluorescence intensities were normalized so that the average green to red ratio of all chromosomes in each metaphase was 1.0. The final results were plotted as a series of green to red ratio profiles and corresponding standard deviation for each human chromosome from p- to q-telomere. At least 12 ratio profiles were averaged for each autosome and 10 for the X chromosome to reduce noise. Green to red ratios greater than 1.2 were considered as gains and ratios less than 0.80 as losses. If the ratio exceeded 2.0, this was considered as a high-level amplification. The cutoff values were based on the average value from the whole material. Heterochromatic regions in the centromeric and paracentromeric parts of the chromosomes, the short arm of the acrocentric chromosomes, the Y-chromosome, as well as the regions next to the telomeres were not included in the evaluation. As an internal control for the CGH methodology, nine of the tumor samples were hybridized and analyzed on two different occasions, giving concordant results in all cases.
Statistical Analyses
Individual chromosome copy number changes between VH3–21-utilizing mantle cell lymphomas and the remaining mantle cell lymphomas were compared using the Fisher's exact test. Correlations between the number of CGH aberrations and the two groups of patients according to tumor VH3–21 usage or not were analyzed by the Mann–Whitney U-test using the StatView 5.0.1 software. P-values below 0.05 were considered significant. The overall survival was calculated from the date of diagnosis until last follow-up or death. Kaplan–Meier survival analysis and log-rank analysis were performed using the Statistica 6.0 software to study the prognostic impact of VH gene usage and P was calculated as two-sided.
Results
CGH Alterations in Mantle Cell Lymphoma
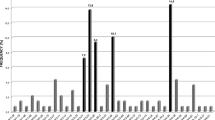
DNA from 37 mantle cell lymphoma patients was screened for numerical chromosomal aberrations using CGH. The deviations found are illustrated in Figure 1 and detailed for each tumor in Table 3. Chromosomal aberrations detected by CGH were found in tumor DNA of 32 of the 37 patients (86%). The most frequent aberrations were gains in 3q (30%), 7p (19%), 8q (16%) and 15q (16%) and losses within 6q (27%), 9p (21%), 9q (30%), 11q (16%) and 13q (51%). Overall, losses were more common compared to gains, and only one high-level amplification (8q) was identified.

CGH profiles showing copy number changes detected in 32 of the 37 mantle cell lymphoma tumors studied. Each line represents one alteration detected in one tumor, with losses indicated to the left and gains to the right of the ideograms. The bold line indicates high-level amplification. The dashed lines represent tendencies to losses that did not reach the threshold of 0.80 (applies only for chromosome 11). In (a) all CGH alterations detected are illustrated. The most commonly involved chromosomes in (b) VH3–21+ tumors and (c) non-VH3–21 tumors, respectively, are shown below. (n) denotes the total number of tumors analyzed in each category.
Generally, the interpretation of the CGH profiles regarding chromosomal alterations was straightforward. However, on the long arm of chromosome 11, six tumors showed typical deletions, while three additional cases showed borderline losses that were close but just short of the cutoff value of 0.80 (Figures 1 and 2). These three cases were thus regarded as having tendencies to loss within 11q but were not included in the statistical analyses.

CGH profiles of mantle cell lymphoma cases with (a) losses, or (b) tendencies to losses involving the long arm of chromosome 11. In addition, case 37 showed a gain encompassing 11q11–14. The mantle cell lymphoma case number is given above, and the deleted region is marked to the left of the corresponding CGH profile. The commonly deleted region was mapped to 11q22, as illustrated next to the ideogram of chromosome 11.
CGH Alterations in Relation to VH Gene Usage
The VH gene rearrangements were previously amplified and sequenced for all 37 mantle cell lymphoma tumors6 and the VH gene usage is detailed in Table 3. Within the group of VH3–21+ tumors (n=14), half as many CGH deviations were found compared to the non-VH3–21 tumors (n=23) (Table 4). Specifically, the VH3–21+ cases exhibited between 0 and 5 aberrations (mean=2.4), while in the non-VH3–21 tumors the CGH imbalances ranged from 0 to 12 (mean=4.9). The lower number of aberrations in the VH3–21+ group was statistically significant (P=0.027, Mann–Whitney U-test).
Among the most commonly identified CGH alterations, different minimal gained or deleted regions were noted between the VH3–21+ and non-VH3–21 groups (Figure 1b and c). For 3q, the minimal gained region was 3q22–qter in the VH3–21+ group compared to 3q25 among the non-VH3–21 samples. Moreover, differences in minimal deleted regions were found within the long arm of chromosomes 6, 9 and 13; 6q25–qter vs 6q25, 9q21 vs 9q21–22 and 13q21 vs 13q33–qter, in VH3–21+ tumors compared to non-VH3–21-utilizing tumors, respectively. Noteworthy, the altered areas were overlapping in 3q, 6q and 9q, while the deleted regions in 13q differed between the two groups.
In Table 4, the most common CGH alterations are listed in relation to VH gene usage. Deletions in 9p were exclusively found among the non-VH3–21 samples. This was also the case for gains in 7p and 15q. The remaining frequent aberrations (gains in 3q and 8q, and losses within 11q and 13q) were also over represented in the non-VH3–21 group, except for losses in 6q and 9q. Among these alterations the gain of 7p and loss of 9q were significantly more frequent, P=0.031 and 0.015, respectively, within the non-VH3–21 group.
Survival Analysis
The Kaplan–Meier curve in Figure 3 shows the overall survival from the time of diagnosis for patients with VH3–21+ tumors (n=14) compared to the remaining patients (n=23). The median overall survival was 37 months compared to 23 months, respectively. Although the difference in median survival was not statistically significant (P=0.13), an obvious trend towards better survival was observed for the VH3–21+ patients. In this cohort of 37 mantle cell lymphomas, there was no survival difference between patients with morphological classical mantle cell lymphoma (n=27) compared to patients with the blastoid variant (n=10), with median overall survival 27 months compared to 24 months, respectively. However, to exclude any possible influence of the morphological subtype on survival between patients with VH3–21+ tumors compared to the remaining patients, we analyzed the survival difference within each of the two morphological subgroups. Within the group of classical mantle cell lymphomas, median survival of VH3–21+ mantle cell lymphoma patients (n=7) was 43 months, compared to 23 months for non-VH3–21 mantle cell lymphoma patients (n=20), reaching statistical significance at P=0.03 (data not shown). Within the group of mantle cell lymphomas with a blastoid variant, only three non-VH3–21 cases were available (compared to seven VH3–21+), whereby further analysis was not applicable.

Kaplan–Meier survival curve comparing VH3–21+ mantle cell lymphoma patients with the mantle cell lymphoma patients utilizing other VH genes, showing a tendency towards better survival for the VH3–21+ group. Open circles represent patients who died and crosses denote censored patients.
Discussion
Mantle cell lymphoma is a disease cytogenetically characterized by a t(11;14), an abnormality that results in cyclin D1 overexpression and a deregulated cell cycle. However, this aberration alone is probably not sufficient for tumor formation15 and knowledge of additional genetic changes in these tumors may therefore be of great importance. Since VH3–21+ mantle cell lymphoma has been suggested to constitute a new subgroup of mantle cell lymphoma with certain molecular characteristics and a tendency towards superior overall survival,6 we characterized the aberration profiles in VH3–21 positive and negative mantle cell lymphoma by CGH. In agreement with previous studies,9, 10, 11, 12 overall gains of 3q, 7p and 8q and losses of 6q, 9p, 11q and 13q were frequently found in this material. Interestingly, differences between the subsets were evident, such as a lower mean number of alterations detected by CGH in VH3–21+ mantle cell lymphoma and skewed distribution of certain aberrations (Figure 1b and c).
The most frequent aberration identified in the present study was a loss of part or all of chromosome 13, detected in 51% of the cases. The deleted region recurrently identified encompassed 13q14–qter, which is in agreement with other studies.9, 10, 11 Interestingly, deletions involving 13q14 are the most frequently occurring chromosomal changes in chronic lymphocytic leukemia,16 a disease that has certain clinical and biological characteristics in common with mantle cell lymphoma. At present, a few candidate genes have been identified in the 13q14 region; however, it is not known if they are disease-related.17, 18 The presence of 13q14 deletions in both chronic lymphocytic leukemia and mantle cell lymphoma may indicate a common genetic component involved in the tumorigenesis of the two malignancies. Another possible mechanism is that different, but closely linked genes may be involved. In the current series, we noted a nonsignificant tendency to overrepresentation of 13q deletions within the group of non-VH3–21 tumors compared to the VH3–21+ tumors. Furthermore, the non-VH3–21 utilizing mantle cell lymphomas had a propensity for more telomeric losses while the VH3–21+ cases encompassed band 13q21 as the minimal deleted region (Figure 1b and c). Noteworthy, the minimal deleted region may in fact encompass the adjacent band 13q14 as well since the proximal limitation to band q21 is determined only by one patient.
Chromosome 9 deletions appeared to be a common finding, which is in agreement with other studies.9, 10, 11, 12 However, in our material, a dual pattern of losses was found in which the short arm was deleted only in the non-VH3–21 mantle cell lymphoma, while losses of 9q were found in both VH3–21+ and non-VH3–21 cases. Of note, the deletions on 9p encompass the INK4a/ARF locus at 9p21 that encodes two tumor suppressors, p16INK4a and p14ARF. Deletions of these genes are recurrent findings in mantle cell lymphoma and it has been reported that these deletions, in combination with cyclin D1 overexpression, have synergistic effects on proliferation.5
In total, 30% of the cases, VH3–21+ as well as non-VH3–21, showed a gain in 3q. This is the most common finding in mantle cell lymphoma in general and is suggested to be an early event in mantle cell lymphomagenesis.9, 10, 11, 12 Overexpression of BCL6, located at 3q27, is usually not found in spite of a 3q gain.10 However, our data suggest a minimal gained region of 3q25, which supports the involvement of another oncogene. Worth mentioning, this more proximal region was extracted from the non-VH3–21 group. Within this region, cancer-relevant genes are sparse, and further investigation may be fruitful.
In our study, 10 cases (27%) showed loss of 6q, relatively equally distributed between the VH3–21+ and non-VH3–21 groups. Partly overlapping regions of minimal deleted region appeared in the two groups; the VH3–21+ tumors showed loss of 6q25–qter, while the non-VH3–21 tumors demonstrated deletions involving 6q25 (Figure 1b and c). A previous study on mantle cell lymphoma has shown deletions encompassing bands 6q25–26.9 Taken together, the available data would place the target region within 6q25. Losses of 6q are common findings in lymphoid malignancies,19 not only in mantle cell lymphomas. Recently, it has been shown that deletions of 6q16.3 are associated with adverse prognosis in follicular lymphoma.20 This indicates that more detailed studies with techniques that allow high-resolution mapping of 6q are of importance. At present, no candidate genes with implied pathological significance are identified within this region.
Another commonly occurring alteration was gain in the short arm of chromosome 7, found in 19% of the cases. All of these gains were found in non-VH3–21 mantle cell lymphomas and not in any of the VH3–21+ cases (P=0.031). However, the effect of the additional 7p genetic material and why this aberration is allocated exclusively to non-VH3–21 mantle cell lymphoma cases remains to be clarified.
At the 11q22 region, six tumors (16%), of equal distribution between VH3–21+ and non-VH3–21 cases, showed clear losses. Moreover, three additional tumors showed CGH profiles with tendencies to deletion, but did not exceed the threshold value for what is considered a true loss (0.80) (Figures 1 and 2). Possibly, this was due to heterozygous deletions or heteroclonality within the neoplasia, whereby the 11q deletions are only present in a fraction of the malignant cells. Chromosomal deletion of 11q22–23 is a common finding in mantle cell lymphoma. This region harbors the DNA damage-sensing ATM (ataxia telangiectasia mutated) gene and it has previously been shown that loss of one allele and mutation of the remaining allele is associated with progressive disease.21 Located in the adjacent chromosomal region, 11 Mb telomeric to ATM, is the gene encoding histone H2AX. This protein is suggested to be a genomic caretaker requiring both alleles for full tumor-protective function.22 Since ATM inactivation does not seem to be found in all mantle cell lymphomas when there is loss of the corresponding region, it is interesting to note that there are more genes with potential tumor-association in that region. Both ATM and histone H2AX are involved in regulating the response to processes during V(D)J recombination. However, whether histone H2AX plays a role in mantle cell lymphoma development is not yet elucidated.
To summarize, significantly fewer CGH aberrations on average were found in VH3–21+ compared to non-VH3–21 mantle cell lymphoma. Considering that additional chromosomal instability is often accompanied by a more adverse clinical course, this may be one explanation why VH3–21+ patients show a tendency to have better prognosis. Interestingly, despite the fact that 50% of the VH3–21+ tumors had blastoid morphology, which is normally associated with worse prognosis, the patients with VH3–21+ tumors had longer median survival. Furthermore, differences in the profiles of aberrations were demonstrated between the groups, since deletion of 9p and gains of 7p and 15q were exclusively found in the non-VH3–21-utilizing tumors. In addition to less chromosomal complexity, the absence of specific aberrations could also be a possible explanation for the superior outcome of VH3–21+ cases. In conclusion, these novel findings reinforce the hypothesis that VH3–21+ tumors constitute an own entity within mantle cell lymphoma and provide some genetic explanation to account for the less aggressive clinical behavior of these mantle cell lymphoma tumors.
References
Campo E, Raffeld M, Jaffe ES . Mantle-cell lymphoma. Semin Hematol 1999;36:115–127.
Thorsélius M, Walsh S, Eriksson I, et al. Somatic hypermutation and V(H) gene usage in mantle cell lymphoma. Eur J Haematol 2002;68:217–224.
Jaffe ES, Harris NL, Stein H, et al (eds). World Health Organization Classification of Tumors. Pathology and Genetics of Tumors of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon: 2001.
Barista I, Romaguera JE, Cabanillas F . Mantle-cell lymphoma. Lancet Oncol 2001;2:141–148.
Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–197.
Walsh SH, Thorsélius M, Johnson A, et al. Mutated VH genes and preferential VH3-21 use define new subsets of mantle cell lymphoma. Blood 2003;101:4047–4054.
Kienle D, Kröber A, Katzenberger T, et al. VH mutation status and VDJ rearrangement structure in mantle cell lymphoma: correlation with genomic aberrations, clinical characteristics, and outcome. Blood 2003; 102:3003–3009.
Bertoni F, Conconi A, Cogliatti SB, et al. Immunoglobulin heavy chain genes somatic hypermutations and chromosome 11q22–23 deletion in classic mantle cell lymphoma: a study of the Swiss Group for Clinical Cancer Research. Br J Haematol 2004;124:289–298.
Allen JE, Hough RE, Goepel JR, et al. Identification of novel regions of amplification and deletion within mantle cell lymphoma DNA by comparative genomic hybridization. Br J Haematol 2002;116:291–298.
Bea S, Ribas M, Hernandez JM, et al. Increased number of chromosomal imbalances and high-level DNA amplifications in mantle cell lymphoma are associated with blastoid variants. Blood 1999;93:4365–4374.
Bentz M, Plesch A, Bullinger L, et al. t(11;14)-positive mantle cell lymphomas exhibit complex karyotypes and share similarities with B-cell chronic lymphocytic leukemia. Genes Chromosomes Cancer 2000;27:285–294.
Monni O, Oinonen R, Elonen E, et al. Gain of 3q and deletion of 11q22 are frequent aberrations in mantle cell lymphoma. Genes Chromosomes Cancer 1998;21:298–307.
Li AH, Rosenquist R, Forestier E, et al. Clonal rearrangements in childhood and adult precursor B acute lymphoblastic leukemia: a comparative polymerase chain reaction study using multiple sets of primers. Eur J Haematol 1999;63:211–218.
Kallioniemi A, Kallioniemi OP, Sudar D, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992;258:818–821.
Lovec H, Grzeschiczek A, Kowalski MB, et al. Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma in transgenic mice. EMBO J 1994;13:3487–3495.
Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–1916.
Migliazza A, Bosch F, Komatsu H, et al. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood 2001;97:2098–2104.
Hammarsund M, Corcoran MM, Wilson W, et al. Characterization of a novel B-CLL candidate gene—DLEU7—located in the 13q14 tumor suppressor locus. FEBS Lett 2004;556:75–80.
Mitelman F, Johansson B, Mertens F, (eds). Mitelman Database of Chromosome Aberrations in Cancer##http://cgap.nci.nih.gov/Chromosomes/Mitelman 2003.
Henderson LJ, Okamoto I, Lestou VS, et al. Delineation of a minimal region of deletion at 6q16.3 in follicular lymphoma and construction of a bacterial artificial chromosome contig spanning a 6-megabase region of 6q16–q21. Genes Chromosomes Cancer 2004;40:60–65.
Schaffner C, Idler I, Stilgenbauer S, et al. Mantle cell lymphoma is characterized by inactivation of the ATM gene. Proc Natl Acad Sci USA 2000;97:2773–2778.
Celeste A, Difilippantonio S, Difilippantonio MJ, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003;114:371–383.
Acknowledgements
We acknowledge Per Anders Flordal for assistance in the statistical processing. We also thank Drs Christer Sundström, Tor Olofsson, Mats Jerkeman, Mats Ehinger, Michael Dictor, Anna Porwit-MacDonald, Erik Björck, Göran Roos and Birgitta Sander for essential contributions of tumor samples and clinical data. This work was supported by the Swedish Cancer Society, the Stockholm County Council, the Swedish Society of Medicine and the Swedish Society for Medical Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Flordal Thelander, E., Walsh, S., Thorsélius, M. et al. Mantle cell lymphomas with clonal immunoglobulin VH3–21 gene rearrangements exhibit fewer genomic imbalances than mantle cell lymphomas utilizing other immunoglobulin VH genes. Mod Pathol 18, 331–339 (2005). https://doi.org/10.1038/modpathol.3800237
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800237
Keywords
This article is cited by
-
Clinical utility of recently identified diagnostic, prognostic, and predictive molecular biomarkers in mature B-cell neoplasms
Modern Pathology (2017)
-
Hypermutation in mantle cell lymphoma does not indicate a clinical or biological subentity
Modern Pathology (2009)
-
Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics
Nature Reviews Cancer (2007)
