
Abstract
In the PCK rat, a rodent model of Autosomal Recessive Polycystic Kidney Disease (ARPKD), a spontaneous splicing mutation of Pkhd1 initiates hepatic cyst development. Cystic cholangiocytes possess short and malformed cilia that do not express fibrocystin, the Pkhd1 protein. During the disease course, cysts continue to grow; however, the mechanisms underlying cyst progression are unclear due in part to the lack of suitable cell lines to study cystogenesis. Here, we describe the development of a PCK-derived cholangiocyte cell line (PCK-CCL). Normal rat cholangiocytes (NRCs) were used as a control. The PCK-CCL maintained a cholangiocyte phenotype as assessed by the expression of the CK-19, CK-7 and GGT. PCK-CCL grown on collagen formed a polarized monolayer with well-developed junctional complexes, and distinct apical and basolateral membranes. Compared to NRCs, cilia in the PCK-CCL were short and malformed and did not express fibrocystin. The PCK-CCL exhibited a higher rate of proliferation (P<0.05) with a doubling time approximately half that of NRCs. By RT-PCR analysis of exons 33–37, an approximately 800 bp product of Pkhd1 was amplified in NRCs. In contrast and as expected, in the PCK-CCL, the Pkhd1 amplicon was smaller (∼630 bp) reflecting the IVS35-2A → T mutation. PCK-CCL and NRCs seeded in 3-D cultures formed cystic structures; however, the PCK cysts expanded progressively up to day 21 while cysts formed by NRCs remained the same size after day 9. In summary, we have developed a cholangiocyte cell line from the PCK rat that retains properties of the cholangiocytes lining hepatic cysts in vivo. The cells have been grown continuously for ∼18 month and 45 passages without crisis or senescence. The morphology and growth characteristics of the PCK-CCL are consistent with those seen in vivo in the PCK rat, suggesting that this cell line will be useful in dissecting the mechanisms of hepatic cyst formation.
Similar content being viewed by others

Main
Autosomal Recessive Polycystic Kidney Disease (ARPKD) is a devastating infantile disease with an incidence of 1:20 000 that often results in fetal or neonatal death due to markedly enlarged kidneys, impaired lung function and pulmonary hypoplasia. Liver cysts develop in most babies who survive the perinatal period and, with age, liver cystic disease becomes the major cause of morbidity and mortality.1, 2, 3, 4, 5 Biliary dysgenesis is characterized by congenital hepatic fibrosis, bile duct dilation and cyst formation. The hepatic cysts are lined by biliary epithelia (ie, cholangiocytes) and increase in number and size throughout life. The gene responsible for ARPKD, PKHD1, encodes a large trans-membrane protein, fibrocystin, whose function is unknown.6, 7, 8 It has been shown recently by us9, 10 and others11, 12 that fibrocystin is expressed in primary cilia of biliary and renal epithelia under normal conditions. Disruption of fibrocystin expression in the PCK rat, an animal model of ARPKD, results in kidney and liver cystogenesis.9 Despite the identification of the genetic defect that causes this disease, the pathophysiology of cyst formation, growth and expansion is still unclear. It has been suggested that abnormalities in cell proliferation, fluid secretion and extracellular matrix biology all contribute to the disease.1, 2, 13
Various animal models of ARPKD have been developed over time, many of them display distinct liver pathology.14, 15, 16 Of these models, the PCK rat closely resembles human ARPKD. The gene, Pkhd1, that causes kidney and liver disease in these rats is orthologous to the human PKHD1.6 We have recently described in detail the morphology of cholangiocytes that line liver cysts in the PCK rat.17 In particular, we have shown that cysts originate from the intrahepatic bile ducts, and that cystic cholangiocytes have a disease specific phenotype including short and malformed cilia that do not express fibrocystin.
Studies of the mechanisms of liver cyst formation in ARPKD have been limited by the lack of suitable cell lines. Thus, to explore the mechanisms of cyst growth and expansion in ARPKD, our aim was to develop a cholangiocyte cell line derived from the intrahepatic bile ducts of the PCK rat which we designated PCK-CCL (ie, PolyCystic Kidney Cholangiocyte Cell Line). We believe that the PCK-CCL will provide a valuable tool to study the molecular mechanisms underlying cyst development and progression in ARPKD.
Materials and methods
Cell Culture of Primary PCK Cholangiocytes
Biliary explants were isolated from 2- to 3-month old male PCK rats as previously described.18 Briefly, the livers were perfused with Hepes-buffer saline (140 mM NaCl, 5.4 mM KCl, 0.8 mM Na2HPO4, 25 mM HEPES, pH 7.4) plus 2 mM CaCl2 and 0.02% collagenase type II and mechanically disrupted to obtain a clean portal tract residue. They were then placed in a solution of 25 ml of RPMI medium, 10 mg hyaluronidase, 8 mg collagenase type XI, and 6 mg DNase and shaken for 30 min in 37°C water bath. These pieces were gravity sedimented and washed with HBS to remove the digestive enzymes. The cells were entered into culture as previously described.19 Biliary explants were washed three times with DMEM/F12 medium, re-suspended in a large volume of DMEM/F12 and placed in a 10 cm tissue culture dish in order to shed contaminant cells such a fibroblasts. After approximately 2 h the explants were re-suspended in 1.5 ml of type I rat tail collagen (1.5 mg/ml, BD Biosciences, Bedford, MA, USA) supplemented with 10% Matrigel (BD Biosciences, Bedford, MA, USA) and evenly spread in a 35 mm dish previously coated with 1 ml of the collagen mixture. The plates were placed in a tissue culture incubator (37°C, 5% CO2, and 100% humidity) for 30 min to allow the gel to solidify. Cholangiocyte media was then poured over the gel and the plates were returned to the incubator. The medium was changed every day to avoid possible contamination. Within 2–3 days of culture, the cholangiocytes proliferated and formed cysts. The medium was removed from these dishes and replaced with DMEM/F12 containing 2 mg/ml collagenase type XI (Sigma) and allowed to digest 45 min at 37°C. Under an inverted scope in sterile conditions, the cysts were dissected using sterile needles and Pasteur pipets to select areas of cholangiocytes free from fibroblast contamination. The cysts were then washed by gravity sedimentation, lightly trypsinized and seeded on top a thick layer of the rat tail collagen. These cells seeded down and proliferated as monolayers across the majority of the plate.
Immunofluorescence Confocal Microscopy
Cholangiocytes grown on collagen coated cover slips were fixed with ice cold methanol for 30 min and permeabilized for 2 min with 0.2% Triton X 100. After blocking, the cells were incubated overnight at 4°C with primary antibodies against CK-7 (1:50, ICN), CK-19 (1:200, Sigma), Vimentin (BioGenex, San Ramon, CA, USA), acetylated apha-tubulin (1:200, Sigma), fibrocystin (gift from Dr Ward), ZO-1 (1:100, Zymed, Carlsbad, CA, USA); Beta-catenin (1:100, BD Biosciences, Bedford, MA, USA). Alexa-488 and Alexa-594 (1:500, Molecular Probes, Carlsbad, CA, USA) was used as a secondary antibody and nuclei were stain with 4′,6-diamidino-2-phenylindole (DAPI). Gamma-glutamyl transpeptidase (GGT) enzyme immuno-histochemical staining was performed on cytospun fractions of the PCK-CCL.
Transmission and Scanning Electron Microscopy
The cell lines were grown to 10 days post-confluence on collagen coated 24-well tissue culture inserts. The cells were fixed in 4% paraformaldehdye/2% gluteraldehyde for 1 h at room temperature or at 4°C overnight, then post-fixed for 1 h in 1% osmium tetroxide. For TEM, the samples were dehydrated, embedded in Spurrs resin, sectioned at 80 nm, and observed using a Joel 12 electron microscope (Joel USA Inc., Peabody, MA, USA). SEM samples were dehydrated, dried in a critical point drying device, mounted, sputter coated, and examined with a Hitachi S-4700 microscope (Hitachi Inc., Pleasanton, CA, USA). Scanning micrographs of PCK-CCL and NRCs were used to measure the length of cilia as we previously described.9
TEER Measurements
The cells were seeded on Falcon 12-well cell culture inserts (BD Biosciences) previously coated with a thin layer of the collagen/Matrigel mixture. The inserts were monitored each day for confluence and electrical resistance using a Millicel-ERS ohm-voltmeter (Millipore Corporation, Bedford, MA, USA). The TEER values were calculated by converting to Ω cm2.
RNA Isolation and RT-PCR
The cell were grown to confluence in 75 cm2 flasks, scraped and washed in PBS. RNA was isolated using TRIzol Reagent (Invitrogen). Pkhd1 mRNA was detected by reverse transcription of the RNA to obtain cDNA, followed by polymerase chain reaction using the SuperScript III kit (Invitrogen). The following primers, corresponding to 5′ and 3′ regions of the Pkhd1 (forward: TGGTGCTCCGTGCCAAGTC, and reverse: GAGTTCCATTCCCTCACAGCCAG) were designed and synthesized by Mayo Core Facility. RT-PCR amplification was performed for 35 cycles. Each cycle consisted of denaturation at 94°C for 1 min, annealing at 56°C for 2 min and extension at 72°C for 3 min.
Cell Proliferation Assay
The PCK-CCL and NRCs were seeded at 96-well plate at density 5000 cells/well. The plates were incubated (37°C, 5% CO2, and 100% humidity) for 48 h prior to the assay. The rate of cell proliferation was determined using CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega).
3-D Culture
The PCK-CCL and NRCs were suspended in 1.5 ml of type I rat tail collagen (1.5 mg/ml, BD Biosciences) supplemented with 10% Matrigel (BD Biosciences) and poured into 35 mm dish previously pre-coated with 1 ml of the collagen. The collagen gel was allowed to solidify for 20–30 min at room temperature and then overlaid with 1.5 ml of medium, which was replaced every other day. The plates were kept in a tissue culture incubator (37°C, 5% CO2, and 100% humidity). Development of cystic structures was assessed by light microscopy from 0 to 21 days in culture. Using Image J software (NIH), the outer diameter of each cyst was determined from the average of two perpendicular measurements. The surface area of each cyst was calculated at each time point. The changes in the rate of cyst growth and expansion were expressed as a fold-increase compared to the basal value (day 1).
Results
Development of the PCK-CCL
Bile duct explants were isolated from 2- to 3-month old PCK rats as described in Methods. Small portions of these explants were placed onto a mixture of type I rat tail collagen+10% Matrigel. Within 2–3 days, they started to proliferate forming large cysts (Figure 1a). The cysts were dissected using sterile needles and Pasteur pipets, and again placed on top of a thick collagen layer. After 5–7 days, the cells had spread out from the plated cysts to form the colonies of uniformly sized polygonal cells across the majority of the plate (Figure 1b). By this time, little fibroblast contamination was apparent. Thus, these colonies were passaged several times in the same manner in order to further reduce contamination with fibroblasts.

Development of the PCK-CCL. Bile duct explants were isolated by enzyme digestion from 2- to 3-month old PCK rats and placed on type I rat tail collagen supplemented with 10% Matrigel. Cells spread from these explants and proliferated forming cysts (a). The individual cysts were selected and sub-cultured; 5–7 days later they formed a monolayer (b) composed of epithelial cells. Magnifications: × 4 (a and b).
Characterization of PCK-CCL
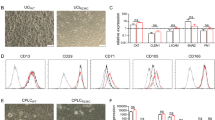
The phenotype of PCK-CCL was determined using a variety of cell-type specific antibodies. The PCK-CCL expressed epithelial cell markers CK-19 (Figure 2a), CK-7 (Figure 2b), and GGT (Figure 2c); <2% of the cells were positive for vimentin, a mesenchymal cell marker (Figure 2d). Thus, the PCK-CCL maintained a cholangiocyte phenotype.

Phenotype of the PCK-CCL. PCK-CCL was stained positively for the epithelial cell markers CK-19 and CK-7 as shown by immunofluorescence confocal microscopy (a and b). Light microscopy demonstrates positive expression of another epithelial cell marker, GGT (c). The majority of the PCK-CCL was negative for mesenchymal cell marker, vimentin (d, immunofluorescence confocal microscopy). Nuclei were stained with DAPI (blue). Magnifications: × 63 (a, b and d), × 20 (c).
We previously reported the establishment of a spontaneously immortalized cholangiocyte cell line derived from normal rats.19 When grown on appropriate matrices, normal rat cholangiocytes (NRCs) form polarized monolayers with suitable electrical resistance and retain the phenotypic and functional characteristics that define these cells in vivo. Thus, we used NRCs for comparative studies between normal and cystic cholangiocytes.
PCK-CCL plated onto a collagen gel spread out as a tight monolayer. Phase-contrast micrograph of the PCK-CCL shows that cholangiocytes were homogenous in size and are polygonal in shape (Figure 3a). The PCK-CCL expressed the tight and adhesion junction markers, ZO-1 (Figure 3b) and B-catenin (Figure 3c), respectively. By transmission electron microscopy, the cells had well differentiated morphology with defined apical and basolateral membranes, abundant microvilli, well-developed junctional complexes, and basolaterally located nuclei (Figure 3d and e). Transepithelial electrical resistance (TEER) in the PCK-CCL was high (Figure 3f).

PCK-CCL growing on the top of collagen gel. Phase-contrast microscopy showed a tight monolayer formed by PCK-CCL grown in culture for 7–14 days (a). Tight junctions were present between adjacent cells as shown by expression of ZO-1 (b). Adhesion junctions were labeled using an antibody against β-catenin (c). Well-maintained polarity is indicated by numerous microvilli, appropriately developed tight (TJ) and desmosomal (D) junctions and basolaterally located nuclei as shown by transmission electron microscopy (d, e). Transepithelial electrical resistance of the PCK-CCL monolayers (circles) was measured during two weeks in culture. NRCs (boxes) were grown under identical conditions and served as a control (f; n=3 for each cell line). Scale bars 10 μm (b and c), magnification × 20 (a).
Cilia in the PCK-CCL
SEM of PCK-CCL (Figure 4a) and NRCs (Figure 4c) grown for 10–14 days under identical conditions revealed that both cell lines developed primary cilia. The result was confirmed by immunofluorescence confocal microscopy (Figure 4b and d). However, cilia in the PCK-CCL were much shorter than in NRCs: 2.59±0.88 μm versus 7.64±1.7 μm, respectively (P<0.05; n=30 for each cell line). By transmission electron microscopy, cilia in the PCK-CCL were malformed (Figure 5a) as compared to NRCs (Figure 5b). SEM images demonstrate aberrant cilia in the PCK-CCL with bulbs on the ciliary tip (Figure 5c). In NRCs primary cilia appear normal (Figure 5d). The observations made in PCK-CCL are consistent with characteristics of cholangiocyte cilia in liver cysts of the PCK rat that we observed in vivo.9, 17

Scanning electron microscopy (a and c; scale bars: 2 μm) and immuno-fluorescence confocal microscopy (b and d; scale bars: 10 μm) of primary cilia in PCK-CCL (a and b) and NRCs (c and d) grown on a collagen gel for 10–14 days after confluence. Cilia were stained with ciliary marker acetylated a-tubulin (red; b and d) and nuclei were stained with DAPI (blue). Inset (a; scale bar 1 μm) shows single primary cilium at higher power.

Transmission (top) and scanning (bottom) electron microscopy of primary cilia in the PCK-CCL and NRCs. In the PCK-CCL, cilia were abnormal (a) and malformed with bulbs on the ciliary tip (c) compared to cilia in NRCs (c and d). Scale bars: 1 μm (a–c).
Expression of Pkhd1 and Fibrocystin in the PCK-CCL
By RT-PCR analysis of exons 33–37, an approximately 800 bp product of Pkhd1 was amplified in NRCs and freshly isolated bile ducts from normal rats. In contrast and as expected, in the PCK-CCL and freshly isolated bile ducts from the PCK rat, the Pkhd1 amplicon was smaller (∼630 bp) reflecting the IVS35-2A → T mutation (Figure 6).6

RT-PCR analysis of Pkhd1 mRNA expression. Level of Pkhd1 mRNA of expected size was detected in PCK-CCL (∼630 bp) and NRCs (∼800 bp). Freshly isolated bile ducts from normal (normal BD) and PCK (PCK BD) rats were used as a positive control.
The expression of fibrocystin in PCK-CCL and NRCs was examined by immunofluorescence confocal microscopy. In NRCs fibrocystin was localized in cilia in addition to the cytoplasm and plasma membrane (Figure 7a–c). In contrast, no fibrocystin was observed in cilia of the PCK-CCL (Figure 7d–f).

Expression of fibrocystin in NRCs (a–c) and PCK-CCL (d–f) by immunofluorescence confocal microscopy. Ciliary marker, acetylated alpha-tubulin is stain in green; fibrocystin is in red; nuclei are stain with DAPI. Scale bars: 10 μm (a–f). Asterisks indicate cilia.
PCK-CCL Growth
Studies of PCK-CCL and NRCs growth were carried out concurrently. The rate of normal and cystic cholangiocytes proliferation was assessed at day 3–7 after the cells were platted. The PCK-CCL showed a higher proliferative response than NRCs; there were statistically significant differences between two cell lines at every time point analyzed (Figure 8). The cell doubling time for NRCs and PCK-CCL was 58.1 and 28.8 h, respectively, indicating that the growth rate of the PCK-CCL was accelerated compared to normal cholangiocytes.

PCK-CCL and NRCs proliferation. The PCK-CCL and NRCs were seeded at density 5000 cells/well. The cells were grown for 2 days prior to the assay. Values are means±s.e. (n=6). *P<0.05.
PCK-CCL Grown in 3-D Culture
The PCK-CCL and NRCs seeded in 3-D culture organized into cystic structures as assessed by light microscopy. In both cell lines, formation of small cysts was observed within 24–48 h and the cysts continue to grow (Figure 9a). Cells were maintained in this configuration for up to 3–4 weeks.

Cysts formed in 3-D culture by PCK-CCL and NRCs. (a): Cysts began to form within a few hours after the cell were seeded. By day 1, small cysts were observed. Cysts continue to grow and expand over time. Scale bars: 500 μm. (b): Quantitation of the surface area of cysts formed by PCK-CCL (boxes) and NRCs (circles). Area measured at day 1 was considered as initial cystic surface area. Cyst dimensions were measured at day 3, 6, 9, 12, 15 and 21 and corresponding changes were expressed as fold increase compared to initial cystic surface area. *P<0.05 as compared to NRCs, n=30 (reflects number of cysts measured for each cell line at every time point analyzed).
Next, we assessed the numbers and size of cystic structures formed by both cell lines in 3-D culture. We did not find any statistically significant differences in number of PCK-CCL and NRCs cysts over 3 weeks of observations. However, the surface areas of cysts formed by PCK-CCL were substantially larger compared to NRCs. The changes in cystic surface area at day 3, 6, 9, 12, 15 and 21 were compared to initial surface area (at day 1) and expressed as fold increase for each cell line. Quantitation of observed changes are shown in Figure 9b. The cystic structures formed by NRCs grew up to days 3–6, around day 9 they reached the plateau and maintained the same size up to the end of experiment. In contrast, PCK-CCL cysts continue to grow and expand progressively up to day 21.
Discussion
Here, we report the development of cholangiocyte cell line designated PCK-CCL, derived from the intrahepatic bile ducts of the PCK rat, an animal model of ARPKD. The cells have been grown continuously for periods exceeding 18 month and over 45 passages without undergoing crisis or senescence suggesting that they become spontaneously immortalized. The PCK cells exhibit a phenotype of abnormal cholangiocytes which line the liver cysts in the PCK rat in vivo in that they: (i) possess shortened, malformed primary cilia; (ii) express mutated Pkhd1 at the mRNA level; (iii) do not express fibrocystin in cilia.
The PCK-CCL can be grown under different conditions: (i) on a single layer of collagen gel when they form a tight polarized monolayer, and (ii) within a collagen/10% Matrigel matrix (ie, 3-D culture) when they form cystic structures. Both experimental systems (cell monolayer and 3-D culture) will be useful to study the mechanisms of cyst formation and role of different factors (derived from apical or basolateral membranes) that contribute to cyst growth and expansion.
The PCK-CCL displays the epithelial characteristics of cholangiocytes as confirmed by expression of the epithelial cell markers CK-7, CK-19 and GGT. When the cystic cells were seeded at a single layer of collagen gel, they spread out as a tight monolayer with a defined apical and basolateral membranes, well developed microvilli and junctional complexes. The PCK-CCL forms electrically tight monolayers. TEER, which is known to reflect cell density and tight junctional integrity20 was actually higher in the PCK-CCL compared to NRCs. The PCK-CCL possesses primary cilia on the apical domain. However, compared to NRCs, PCK-CCL cilia were structurally (significantly shorter and malformed) and functionally (they did not express fibrocystin) abnormal. It has been shown previously that the gene, Pkhd1, is responsible for the cystic phenotype in the liver and kidneys of the PCK rat. The disease is a result of a spontaneous splicing mutation IVS35-2A → T.6 By employing RT-PCR, we showed the differences in the expression of Pkhd1 mRNA between normal and cystic cholangiocytes, further supporting that the PCK-CCL maintains the phenotype seen in vivo. Finally, PCK-derived cholangiocytes proliferate more rapidly than NRCs. All these observations suggest that the cystic phenotype is maintained in established PCK-CCL.
When the PCK cholangiocytes were grown in 3-D culture, they formed spherical cystic structures that expanded in size over time. The 3-D culture system was first established in 1979 using mammary carcinoma tissue.21, 22 It has been applied widely to many organs and tissue.23 For example, different kidney epithelial cell lines are known to form spherical cysts within collagen gel.24, 25, 26 Bile duct epithelial cells and gallbladder-derived cells are also able to form cystic structure in 3-D culture.23, 27, 28, 29 Thus, The PCK-CCL cultured between two layers of collagen/Matrigel is an excellent model to study cystogenesis in vitro. The 3-D experimental system will allow us to evaluate and test the role of potential factors (activators and inhibitors) that contribute to cyst progression.
Different hormones, growth factors, cytokines (individually or in combination) could promote cyst expansion. Studies of the mechanisms of liver cysts progression in ARPKD have been hampered by the lack of available continuous cyst-derived cell lines. Several investigators have used cyst-derived cells in primary culture. As reported recently by Sato and coauthors, primary culture of bile duct epithelial cells derived from the PCK rat has been employed to study pathogenesis of ARPKD.30 These cells maintain features of cholangiocytes and had been passaged four times, however, the further fate of these cells in culture is unknown.
Our spontaneously immortalized PCK-CCL derived from the biliary tree of an animal model of APRKD will provide a useful an in vitro system to study the mechanisms of cyst growth and expansion. Having in our disposition two cell lines (one with normal and one with disease phenotype), that can be grown in a single gel (forming monolayer) or in 3-D culture (forming cysts) will help to dissect the differences in cellular response by normal and cystic cholangiocytes to many factors and stimuli involved in cell proliferation, secretion and cell-matrix interactions, processes that underlying liver cystogenesis.
References
Wilson PD . Polycystic kidney disease: new understanding in the pathogenesis. Int J Biochem Cell Biol 2004;36:1868–1873.
Davenport JR, Yoder BK . An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol 2005;289:F1159–F1169.
Zerres K, Rudnik-Schoneborn S, Steinkamm C, et al. Autosomal recessive polycystic kidney disease. J Mol Med 1998;76:303–309.
Sessa A, Righetti M, Battini G . Autosomal recessive and dominant polycystic kidney diseases. Minerva Urol Nefrol 2004;56:329–338.
Shneider BL, Magid MS . Liver disease in autosomal recessive polycystic kidney disease. Pediatr Transplant 2005;9:634–639.
Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 2002;30:259–269.
Onuchic LF, Furu L, Nagasawa Y, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet 2002;70:1305–1317.
Nagasawa Y, Matthiesen S, Onuchic LF, et al. Identification and characterization of Pkhd1, the mouse orthologue of the human ARPKD gene. J Am Soc Nephrol 2002;13:2246–2258.
Masyuk TV, Huang BQ, Ward CJ, et al. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology 2003;125:1303–1310.
Ward CJ, Yuan D, Masyuk TV, et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet 2003;12:2703–2710.
Menezes LF, Cai Y, Nagasawa Y, et al. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int 2004;66:1345–1355.
Wang S, Luo Y, Wilson PD, et al. The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. J Am Soc Nephrol 2004;15:592–602.
Perrone RD, Grubman SA, Rogers LC, et al. Continuous epithelial cell lines from ADPKD liver cysts exhibit characteristics of intrahepatic biliary epithelium. Am J Physiol 1995;269:G335–G345.
Guay-Woodford LM . Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol 2003;285:F1034–F1049.
Sanzen T, Harada K, Yasoshima M, et al. Polycystic kidney rat is a novel animal model of Caroli's disease associated with congenital hepatic fibrosis. Am J Pathol 2001;158:1605–1612.
Moser M, Matthiesen S, Kirfel J, et al. A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD). Hepatology 2005;41:1113–1121.
Masyuk TV, Huang BQ, Masyuk AI, et al. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol 2004;165:1719–1730.
Ishii M, Vroman B, LaRusso NF . Isolation and morphologic characterization of bile duct epithelial cells from normal rat liver. Gastroenterology 1989;97:1236–1247.
Vroman B, LaRusso NF . Development and characterization of polarized primary cultures of rat intrahepatic bile duct epithelial cells. Lab Invest 1996;74:303–313.
Madara JL . Tight junction dynamics: is paracellular transport regulated? Cell 1988;53:497–498.
Yang J, Guzman R, Richards J, et al. Primary culture of human mammary epithelial cells embedded in collagen gels. J Natl Cancer Inst 1980;65:337–343.
Yang J, Richards J, Bowman P, et al. Sustained growth and three-dimensional organization of primary mammary tumor epithelial cells embedded in collagen gels. Proc Natl Acad Sci USA 1979;76:3401–3405.
Nakanuma Y, Katayanagi K, Kawamura Y, et al. Monolayer and three-dimensional cell culture and living tissue culture of gallbladder epithelium. Microsc Res Tech 1997;39:71–84.
McAteer JA, Dougherty GS, Gardner Jr KD, et al. Scanning electron microscopy of kidney cells in culture: surface features of polarized epithelia. Scan Electron Microsc 1986;(Pt3):1135–1150.
Sakurai H, Nigam SK . In vitro branching tubulogenesis: implications for developmental and cystic disorders, nephron number, renal repair, and nephron engineering. Kidney Int 1998;54:14–26.
Jiang ST, Liao KK, Liao MC, et al. Age effect of type I collagen on morphogenesis of Mardin-Darby canine kidney cells. Kidney Int 2000;57:1539–1548.
Katayanagi K, Kono N, Nakanuma Y . Isolation, culture and characterization of biliary epithelial cells from different anatomical levels of the intrahepatic and extrahepatic biliary tree from a mouse. Liver 1998;18:90–98.
Ishida Y, Smith S, Wallace L, et al. Ductular morphogenesis and functional polarization of normal human biliary epithelial cells in three-dimensional culture. J Hepatol 2001;35:2–9.
Mano Y, Ishii M, Kisara N, et al. Duct formation by immortalized mouse cholangiocytes: an in vitro model for cholangiopathies. Lab Invest 1998;78:1467–1468.
Sato Y, Harada K, Kizawa K, et al. Activation of the MEK5/ERK5 cascade is responsible for biliary dysgenesis in a rat model of Caroli's disease. Am J Pathol 2005;166:49–60.
Acknowledgements
This work was supported by the National Institutes of Health (Grant DK 24031), by the PKD Foundation and by the Mayo Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Muff, M., Masyuk, T., Stroope, A. et al. Development and characterization of a cholangiocyte cell line from the PCK rat, an animal model of Autosomal Recessive Polycystic Kidney Disease. Lab Invest 86, 940–950 (2006). https://doi.org/10.1038/labinvest.3700448
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700448
Keywords
This article is cited by
-
Applications of organoids in regenerative medicine: a proof-of-concept for biliary injury
Nature Reviews Gastroenterology & Hepatology (2021)
-
NEDD4-family E3 ligase dysfunction due to PKHD1/Pkhd1 defects suggests a mechanistic model for ARPKD pathobiology
Scientific Reports (2017)
-
Demonstration of primary cilia and acetylated α-tubulin in fish endothelial, epithelial and fibroblast cell lines
Fish Physiology and Biochemistry (2016)
-
Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis
Laboratory Investigation (2014)
-
Immortalized liver endothelial cells: a cell culture model for studies of motility and angiogenesis
Laboratory Investigation (2010)
