
Abstract
Given evidence for excessive striatal dopamine activity in schizophrenia, we sought to test the hypothesis that dopaminergic innervation in the striatum is abnormally elevated, and a secondary hypothesis that age-related loss is accelerated. Twelve schizophrenic subjects on stable doses of medications, along with 12 age and sex-matched healthy control subjects, underwent positron emission tomography (PET) studies with [11C]dihydrotetrabenazine (DTBZ), which binds to the vesicular monoamine transporter, type 2 (VMAT2). DTBZ binding reflects principally dopaminergic projections in the striatum and appears in animal models, over treatment periods as long as two weeks, not to be regulated by antipsychotic drugs. Using an equilibrium analysis, we obtained measurements of the binding potential (BP) of [11C]DTBZ, as well as a transport (K1) measure, corresponding to regional cerebral blood flow. BP in the striatum showed no difference between the patient and control groups, and no differential effect of age. We did not find evidence supporting the hypothesis that excessive dopamine activity in schizophrenia could be explained by increased density of striatal dopamine terminals.
Similar content being viewed by others

Main
Since the 1960's, indirect evidence has implicated dopamine systems in schizophrenia and psychosis (Angrist et al. 1974; Creese et al. 1976; Seeman 1987; Snyder 1972). In the last 10 years, neuroimaging studies targeting dopamine systems have begun to characterize the role of dopamine in schizophrenia, although many questions remain. In vivo studies using radioligands which bind to post-synaptic dopamine receptors have provided equivocal evidence for abnormalities of striatal receptors. Some investigators have reported greater receptor density (Gjedde et al. 1996; Wong et al. 1986), whereas many groups have failed to replicate these findings (Farde et al. 1990; Hietala et al. 1994; Nordstrom et al. 1995; Pilowsky et al. 1994).
A more successful approach has been to measure the change in radioligand binding after the manipulation of synaptic dopamine concentration. Two groups have demonstrated that in response to amphetamine, patients with schizophrenia displace more dopamine D2-ligand than healthy subjects (Abi-Dargham et al. 1998; Breier et al. 1997; Laruelle et al. 1996). Additional evidence in support of increased dopamine activity comes from demonstrations of increased metabolism of [18F]fluorodopa (Dao-Castellana et al. 1997; Hietala et al. 1995; Reith et al. 1994) and [11C]L-DOPA (Lindstrom et al. 1999). The prevailing interpretation of these findings is that schizophrenic patients episodically release more dopamine (Breier et al. 1997; Laruelle et al. 1996), possibly in connection with up-regulated enzyme activity of dopa-decarboxylase induced by low tonic activity of dopaminergic neurons (Reith et al. 1994). Given the importance of this data to the pathophysiology of schizophrenia, alternative explanations need to be ruled out. For instance, schizophrenic patients may have greater dopaminergic innervation in the striatum. Regulation of transmitter release, and uptake, could be normal, while greater terminal density could lead to more release of dopamine after re-uptake blockade and vesicular release by amphetamine.
Although post-mortem studies have found normal density of presynaptic dopamine markers, such as the dopamine transporter (Hirai et al. 1988; Joyce et al. 1988; Pearce et al. 1990) and expressed mRNA for tyrosine hydroxylase (Ichinose et al. 1994), excessive innervation could be obscured by normal aging and more rapid degeneration of terminals in schizophrenia. Dopamine receptors (Seeman et al. 1987; Volkow et al. 1996b), transporters (Volkow et al. 1996a), and vesicles (Frey et al. 1998) all decline with aging, with rates estimated at 2–8% per decade. In the cortex of schizophrenic brains, Hitri and coworkers (1995) demonstrated a steeper age-related loss of dopamine transporter in a post-mortem study. There are reasons to suspect that possible neurotoxic effects of psychosis might also damage striatal dopaminergic neurons. Hyperdopaminergic states induced by amphetamine damage dopaminergic neurons in animal models (Seiden et al. 1976; Villemagne et al. 1998; Wagner et al. 1980) and living humans with a history of stimulant abuse have exhibited reduced binding to the dopamine transporter even after several years of abstinence (McCann et al. 1998). Glutamatergic animal models of schizophrenia using phencyclidine also demonstrate neurotoxic effects on striatal neurons (Jentsch et al. 1998; Mitchell et al. 1998; Sonsalla et al. 1989). Thus, it is plausible that schizophrenia might begin with a greater density of dopaminergic neurons, only to experience accelerated loss with aging.
We report here a test of the hypothesis that schizophrenic patients have greater dopaminergic innervation of the striatum. As a secondary hypothesis, we also tested the prediction of accelerated loss of neurons with aging. We employed a new pre-synaptic marker, [11C](+)dihydrotetrabenazine (DTBZ), which labels the terminal projections of dopaminergic, as well as other monaminergic, neurons (DaSilva and Kilbourn 1992; Henry and Scherman 1989). This compound binds to the vesicular monoamine transporter, type 2 (VMAT2), and can be quantified with positron emission tomography (PET) (Koeppe et al. 1996; Vander Borght et al. 1995b). VMAT2 transports intracellular monoamines into the synaptic vesicles and is expressed in all monoaminergic neurons. In the striatum, 95% of the specific binding represents dopaminergic neurons. Therefore, quantitative measurement of DTBZ uptake in the striatum reflects primarily dopaminergic innervation, permitting us to test the hypothesis.
METHODS
Subjects
Twelve schizophrenic subjects were recruited from a university-staffed community mental health center to participate in the study. Patients underwent a Structured Clinical Interview for Diagnosis (First et al. 1996) to establish a diagnosis of schizophrenia according to DSM-IV criteria (American Psychiatric Association 1994). Demographic information on patient and control subjects are listed in Table 1 . Two patients had prior diagnoses of alcohol and substance abuse (including one later discovered to have used cocaine), which were in remission for more than 5 years. One patient had a prior history of depressive disorder not otherwise specified, in remission at the time of the PET study. All patients were outpatients on stable doses of antipsychotic medication at the time of the scan and were free of significant medical or neurologic illness. Adjunctive medications, with the number of patients indicated in parentheses, included anafranil (1), venlafaxine (1), cogentin (1), clonazepam (2), valproic acid (1), and paroxetine (1). Symptoms were assessed with the 18-item Brief Psychiatric Rating Scale (Overall and Gorham 1962) and the Scale for the Assessment of Negative Symptoms (Andreasen 1983).
Healthy control subjects were recruited from community advertisements. To be eligible, control subjects had to be nonsmokers, without serious medical/neurological illness, not taking medication, and free of all Axis I psychiatric disorders according to a Structured Clinical Interview for Diagnosis (First et al.1996). Twelve subjects were matched to the age and gender of the schizophrenic subjects. The purpose and risks of the study were explained to all subjects, who gave written informed consent to participate in the protocol, as approved by the local institutional review board and the committee on human use of radioisotopes, in accordance with the Helsinki Declaration of 1975.
Positron Emission Tomography
The distribution of radioligand was measured by a Siemens-CTI ECAT EXACT-47 tomograph, which acquires 47 contiguous 3.375-mm thick slices over a 15.8 cm axial field of view. All subjects underwent catheterization of a radial artery and a contralateral antecubital vein and had four porous beads (∼1 mm diameter) labeled with approximately 40 MBq (∼1 mCi) [11C]DTBZ affixed around the scalp to provided fiducial markers for scan re-alignment. Subjects were scanned in a supine position, with eyes and ears unoccluded and room lights dimmed. Using methods described previously (Jewett et al. 1997), 600 ± 60 MBq (16 ± 1.6 mCi) of [11C](+)DTBZ were prepared, containing less than 50 mg mass. The scan began with a bolus injection of half the dose over 30 sec, with the remaining half infused over the subsequent 59.5 min of the 60 min scanning period. Each scan consisted of a four-frame sequence in 3-D mode, with septa retracted, from 0 to 4 min after injection, and then three 10-minute frames from 30 to 60 min.
Heparinized arterial blood samples were collected at 10-sec intervals for 2 min, followed by collection at progressively longer intervals. Plasma was separated from red cells and counted in a NaI well-counter. The plasma radioactivity time course was corrected for radiolabeled metabolites using a rapid Sep-Pak C18 cartridge (Waters, Milford, MA) chromatographic technique similar to that reported for scopolamine and flumazenil (Frey et al. 1992).
Scans were reconstructed with a Hanning filter with a cutoff frequency of 0.5 cycles/ray, with attenuation correction from a 2D transmission scan. The dynamic sequence of scans was corrected for any movement occurring during the acquisition sequence by means of the fiducial scalp markers (“beads”). After reconstruction and attenuation correction, automated algorithms located the center of mass of the beads in a reference frame, and all the other frames in the acquisition sequence were brought into alignment with the fiducial beads (three rotational and three translational degrees of freedom).
Analysis
A pixel-by-pixel equilibrium analysis of scan data yielded the VMAT2 binding site density, expressed as the tissue-to-blood tracer distribution volume (DV), obtained by dividing the concentration of [11C]DTBZ measured by PET by the metabolite-corrected arterial plasma concentration, both taken after equilibrium conditions were obtained (30–60 min after tracer injection). Following estimation of DV, the rate of tracer delivery to tissue (K1), reflecting cerebral blood flow, was calculated from the 0–4-min scan using the estimated DV and an “autoradiographic” approach (Koeppe et al. 1997). Prior work has established that for reversible ligands, the DV parameter, reflecting specific binding, non-specific binding, and free ligand, reliably reflects receptor density (Frey et al. 1991; Koeppe et al. 1994). Moreover, analysis of [11C]DTBZ distribution with a three compartment model shows comparable fidelity, and both two and three compartment models demonstrate excellent correlation with the known distribution of VMAT2 sites in humans (Koeppe et al. 1997).
We tested for group differences using two analytic approaches. In the first approach, we used atlas-defined regions of interest (ROI) applied to anatomically-standardized images. Using the K1 parametric image for each subject, automated algorithms identified over 200 cortical and sub-cortical landmarks, which where then submitted to a three-dimensional, thin-plate spline algorithm (Minoshima et al. 1994). In this step, each individual brain was oriented and warped to Talairach atlas space (Talairach and Tournoux 1988). The entire process occurred without operator intervention or bias, and a visual check of image fidelity ensured that the programs did not make gross errors in image transformation. Since the K1 and DV images for each subject were sampled from the same anatomic space and in excellent registration after alignment via fiducial beads, the transformation matrix derived for each K1 image was applied to each DV image, orienting the DV image in standardized Talairach space (Talairach and Tournoux 1988). Spherical ROIs (11.25 mm diameter) were placed at the bilateral stereotactic coordinates of the caudate (x = ± 9 mm, y = 12 mm, z = 7 mm), posterior putamen (±23, −6, 7), anterior putamen (±21, 6, 7), and ventral striatum (±8, 10, −7). We also placed ROIs (13.5 mm diameter) at the coordinates of the right and left thalamus (±10, −12, 12) and the hypothalamic region (0, −5, −8). Mean values were sampled from the DV and K1 images for each subject.
To reduce variance, we normalized DV by dividing the dienchephalic and striatal DV values by the DV sampled from a 13.5 mm ROI placed over the occipital cortex (0, −78, 10). Subtracting normalized DV in the occipital cortex (equal to 1) from normalized DV in our regions of interest yielded a measure of specific binding, or binding potential (BP). Using the inactive (-)DTBZ isomer, which shows a thousand-fold less affinity for VMAT2 compared to the (+)-isomer, we have evaluated the accuracy of occipital DV as an index of non-specific binding. Based on comparisons with the (-)-isomer, binding of the (+)-isomer in the occipital cortex tends to slightly underestimate specific binding in the putamen by 2% and in the caudate by 7% (Koeppe et al. 1999).
In the second analysis, we used the pixel magnitude of the DV images to identify the striatum, where binding is 2–3-fold greater than cortex. This method did not introduce any anatomic deformation via the K1 image or require operator-drawn ROIs. Using the pixel histograms of the DV image from each subject, automated routines bisected the width of the histogram at 50% of the modal pixel count. Based on prior experience with DTBZ studies from normal and Parkinsonian brains acquired in our center, we have identified a reliable threshold value which corresponds to pixels in the striatum. This threshold was 110% of the value identified by bisecting the pixel histogram (Koeppe et al., unpublished data). From the sum of the pixels above this threshold, we obtained a measure of total binding in the striatum. Based on our work with the inactive (-)DTBZ isomer (Koeppe et al. 1999), we assumed that non-specific binding in the striatum would equal the binding in the cortex; therefore, we subtracted this value from each pixel in the striatum and figured a sum equal to non-specific binding. Dividing the total by the non-specific binding and subtracting one yielded a measure of BP.
The data were examined to ensure normality, and where non-normal distributions were found, non-parametric statistics were employed. Group comparisons were performed by unpaired t-tests and mixed-model ANCOVA, using age as a covariate. Statistical analysis was performed with SysStat statistical software (SysStat, Inc., Evanston, IL).
RESULTS
ROI Analysis
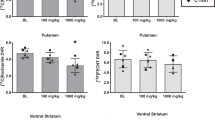
Analysis of the K1 and BP data for right and left hemispheres showed no significant differences between hemispheres; therefore, we have combined the bilateral ROIs for the following analysis. The K1 data (Figure 1A), reflecting cerebral blood flow, showed no group differences, except for reduced flow in the region of the diencephalic nuclei for the patient group (t = 2.29, df = 22, p = .03). We examined the quality of equilibrium achieved, by measuring the percentage change of the raw DV measure across the three 10-min frames, acquired 30–60 min after injection. This was +4.2 ± 6.4% for the striatum, or approximately 8% per hour. The slope of this change did not differ between the groups.

This figure shows (A) K1, or tracer transport, and (B) binding potential (BP) in a priori regions of interest for schizophrenic subjects (SCZ) and healthy control subjects (NORM). * p < .03. There were no significant group differences for BP. CAU: caudate; aPUT: anterior putamen; pPUT: posterior putamen; vSTR: ventral striatum; HYP: hypothalamus; THAL: thalamus; OCC: occipital cortex
There were no significant BP differences between the groups for any region (Figure 1B). We also performed an analysis of covariance with age as a co-variate. As reported in previous work (Frey et al. 1998), we found correlations, at a trend level, between increasing age and decreasing BP in the striatum (Caudate = 1.64–0.012 *[age], r = 0.38, p = .07; posterior putamen = 1.75–0.011*[age], r = 0.34, p = .10). This corresponded to an average 6% decrease in BP per decade of life from 20 to 60. We did not find any differences between the groups in this relationship to age, and as Figure 2 shows for the posterior putamen, the regression slopes between the two groups were very similar. When age was entered as a co-variate in the analysis, the groups still did not differ.

BP in the posterior putamen regressed on age. There were no significant differences between the groups
We also analyzed the raw DV measure in the occipital reference region and found that the patients had a significantly lower DV (mean 4.89 ± S.D. 0.64) compared to the healthy control subjects (5.92 ± 0.89; t = 3.23, df = 22, p = .004). Across all ROIs, we noted similar reductions in DV from 16 to 22%. In order to see if this might affect the relationship between age and specific binding, we performed correlations between age and DV in the occipital region. There were no significant correlations for each group, or in the analysis of both groups combined (r < 0.22).
After completion of the study, we found out that one subject had a prior history of cocaine abuse, which could result in premature loss of dopaminergic neurons (Seiden et al. 1976; Villemagne et al. 1998; Wagner et al. 1980). While this patient had the second-lowest BP values in the striatum, he was also the oldest schizophrenic subject. The results did not change whether or not we excluded him.
Correlations between VMAT2 Binding an Clinical Data
In an exploratory analysis, we tested for correlations between clinical variables and VMAT2 binding. We found no significant relationships between our measure of specific VMAT2 binding in any region and either duration of illness, number of hospitalizations, total symptoms, positive symptoms or negative symptoms. Although a previous report from our center has suggested a relationship between smoking and VMAT2 binding (Gilman et al. 1998), the analysis of a much larger database has not confirmed the initial findings (Koeppe et al., in preparation). Nevertheless, since eight of our schizophrenic subjects smoked, compared to none of the control subjects, we performed Spearman Rank correlations between pack-year history and striatal binding and found no significant correlations.
Pixel Magnitude Analysis
Examining the relative pixel magnitudes, which isolated striatal values, yielded a similar result. The DV threshold (110% of mean) for the control group was 6.13 ± 1.14 (mean ± S.D.) compared to 5.25 ± 0.66 for the schizophrenic group. This was a significant difference (t = 2.30, df = 22, p = .03), and matched our finding across all regions in the ROI analysis. This threshold identified 6020 ± 703 striatal pixels in control subjects and 6243 ± 1230 striatal pixels in the schizophrenic groups. The more specific measures of binding potential revealed no difference between the groups: 0.61 ± 0.07 for the control subjects and 0.61 ± 0.10 for the schizophrenic subjects (t = 0.06 df = 22, p = .99).
DISCUSSION
Studies demonstrating greater displacement of dopaminergic ligands following amphetamine challenge and increased [18F]DOPA uptake (see above) have been put forth as evidence in support of hyperdopaminergic function in schizophrenia. Our findings suggest that an excessive number of dopamine-containing vesicles cannot explain these. Using a pre-synaptic marker of dopaminergic innervation of the striatum, we did not find evidence that dopamine terminal density was greater in our schizophrenic subjects. Likewise, analysis of covariance did not support the hypothesis that patients lost innervation faster than healthy controls, and correcting our measurement for age did not change the findings. This study represents the first application of this marker of dopaminergic innervation to living schizophrenic patients and therefore needs to be considered preliminary; however, the results do have implications for consideration of the dopamine system in schizophrenic pathophysiology.
Although our patients were all taking antipsychotic medication at the time of PET scanning, medication probably did not affect our findings. Studies performed in animals show no effect on vesicle synthesis after 2 days of dopamine agonist and antagonist treatment (Naudon et al. 1994). After 14 days of haloperidol treatment, which clearly induces prominent D2 up-regulation, no change occurs in VMAT2 binding (Vander Borght et al. 1995a). Research into medication effects on VMAT2 regulation has not been exhaustive, which is to say that we are not aware of studies examining long-term medication effects, effects of atypical antipsychotics (most of our patients were taking clozapine), and effects of other psychotropic medications. It is possible that chronic therapy with D2,3,4/5-HT2 antagonists could affect VMAT2 expression or ligand binding. Furthermore, small changes in VMAT2 binding, not detected in the study by Vander Borght et al. (1995a), might have occurred in our patients. In spite of these caveats, the failure to demonstrate regulation of VMAT2 by medication when post-synaptic receptors show very clear changes suggests relative insensitivity to medication status. This stands in contrast to another presynaptic dopamine marker, such as [18F]DOPA (Hietala et al. 1995; Reith et al. 1994), which reflects activity of DOPA decarboxylase, a synthetic enzyme susceptible to negative feedback regulation via the D2 autoreceptor (Cumming et al. 1995; Young et al. 1993).
The absence of a positive finding always raises the question of a type II error, due to sample bias or inadequate experimental power. The BP graph in Figure 1 shows nominally higher values for the schizophrenic subjects (4% greater). However, this magnitude of difference does not approach the increases observed for fluorodopa metabolism, from 16 to 40% in groups with smaller sample sizes (Hietala et al. 1995; Reith et al. 1994). Inspection of the scattergram does show one young schizophrenic subject with a BP nearly 25% higher than the highest control subject, suggesting that a larger sample of young schizophrenic subjects might reveal a significant difference. However, when we examined more subjects from our database of healthy controls, we did find young, healthy subjects with BPs identical to the one schizophrenic subject. Furthermore, in the pixel-magnitude analysis, we found identical values for binding potential in the striatum. In short, we do not believe that studying more subjects would lead to significant differences between the groups.
Somewhat unexpectedly, we noted significantly lower total DV values (free, specific, and non-specific binding) in the pixel magnitude analysis and the region of interest analysis, even in the striatum. This might reflect important structural differences between the patient and control brains. For instance, a reduction in non-specific binding throughout the cortex might be expected from observations of reduced neuropil in schizophrenia (Selemon et al. 1995). Although there are reports of reduced dopaminergic and catecholaminergic innervation of cortex in schizophrenia (Akil et al. 1999; Hirai et al. 1988; Wise and Stein 1973), no more than 6% of [11C]DTBZ total DV constitutes specific binding. Therefore, we cannot attribute the observed 16–22 % decreases in total DV to decreases in specific binding. Partial volume effects in schizophrenic patients, who show global reductions in gray matter (Lauriello et al. 1997; Lim et al. 1996; Zipursky et al. 1992), could also explain this result. However, we note that the K1 measures in the striatum did not differ between the groups, suggesting that partial volume effects did not affect striatal measurements. This agrees with structural images using MRI, which, if anything, show greater volume of striatal structures, such as the caudate, in schizophrenia during antipsychotic treatment (Corson et al. 1999; Jernigan et al. 1991).
Since DV represents a ratio of brain:blood concentration, the reduced total DV throughout the brain could represent differences in the blood concentration. Differences in protein binding, secondary to medications such as clozapine which have high affinity for serum proteins, might have affected the DV measure, although such a reduction in available binding sites would tend to increase DV in the patients. On the other hand, increased serum proteins could cause an apparent decrease in DV, although there is little reason to suspect that this should occur in our patients.
The observation of apparently lower total DV for the patients in the reference region requires comment. Changes in this measure could bias our striatal index of specific binding. The lack of a correlation between age and occipital DV suggests that this measure would not introduce spurious relationships between age and striatal VMAT2 binding. A related concern is that the reduced DV in the occipital reference region could reflect different regional interactions in binding to non-specific proteins, differences in clearance rates or differences in the presence of free tracer. However, the overall pattern of DV measurements, outside the striatum, did not differ between the groups, suggesting that regional interactions were unlikely. Tissue loss in the occipital cortex could cause greater partial volume effects and a reduced total DV. However, this error would tend to drive BP up for the patients. Such a bias would favor our hypothesis that the patients had greater dopaminergic innervation; therefore, it is unlikely to be an explanation of our negative results. Furthermore, our anatomic warping procedure matched all brain sizes to a standardized template, which would attenuate the effects of reduced gray matter. In the interest of an unbiased estimate of dopamine terminals in the striatum, we employed the pixel magnitude analysis, which did not rely on any anatomic warping. This procedure essentially exploited the 3-fold greater signal in striatum, relative to cortex, and normalized to the entire cortical value. Lastly, our in vivo data agree with post-mortem data, using [3H]DTBZ. With non-specific binding in the striatum determined after washing with unlabeled DTBZ, binding potentials in a sample of 14 schizophrenic brains did not differ from matched control tissue (Frey and Meador-Woodruff, unpublished data). Therefore, while questions remain about the interpretation of reduced total DV in the occipital cortex, this uncertainty should not diminish the significance of our demonstration of the absence of increased dopaminergic innervation of the striatum in schizophrenia.
In conclusion, we have demonstrated the first application of a ligand to VMAT2 in schizophrenia, which has served as an index of striatal dopaminergic innervation in other patient groups. The failure to find evidence for increased innervation in a cross-sectional sample of stable, medicated schizophrenic subjects needs to be regarded as preliminary, but still suggestive as to what abnormal dopamine systems might not be in schizophrenia. With relative insensitivity to medication status, it may play a role in schizophrenia research as a longitudinal probe to assess interactions between aging, treatment response and neurite change.
References
Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, van Dyck CH, Charney DS, Innis RB, Laruelle M . (1998): Increased striatal dopamine transmission in schizophrenia: Confirmation in a second cohort. Am J Psychiatry 155: 761–767
Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR, Lewis DA . (1999): Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry 156: 1580–1589
American Psychiatric Association. (1994): Diagnostic and Statistical Manual of Mental Disorders, 4th ed. (DSM-IV) Washington, DC, American Psychiatric Association
Andreasen NC . (1983): The scale for the Assessment of Negative Symptoms. Iowa City: The University of Iowa.
Angrist B, Sathananthan G, Wilk S, Gershon S . (1974): Amphetamine psychosis: Behavioral and biochemical aspects. J Psychiatr Res 11: 13–23
Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D . (1997): Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci U S A 94: 2569–2574
Corson PW, Nopoulos P, Miller DD, Arndt S, Andreasen NC . (1999): Change in basal ganglia volume over 2 years in patients with schizophrenia: Typical versus atypical neuroleptics. Am J Psychiatry 156: 1200–1204
Creese I, Burt DR, Snyder SH . (1976): Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192: 481–483
Cumming P, Ase A, Diksic M, Harrison J, Jolly D, Kuwabara H, Laliberte C, Gjedde A . (1995): Metabolism and blood-brain clearance of L-3,4-dihydroxy-[3H]phenylalanine ([3H]DOPA) and 6-[18F]fluoro-L-DOPA in the rat. Biochem Pharmacol 50: 943–946
Dao-Castellana MH, Paillere-Martinot ML, Hantraye P, Attar-Levy D, Remy P, Crouzel C, Artiges E, Feline A, Syrota A, Martinot JL . (1997): Presynaptic dopaminergic function in the striatum of schizophrenic patients. Schizophr Res 23: 167–174
DaSilva JN, Kilbourn MR . (1992): In vivo binding of [11C]tetrabenazine to vesicular monoamine transporters in mouse brain. Life Sci 51: 593–600
Farde L, Wiesel FA, Stone ES, Halldin C, Nordstrom AL, Hall H, Sedvall G . (1990): D2 dopamine receptors in neuroleptic-naive schizophrenic patients. A positron emission tomography study with [11C]raclopride. Arch Gen Psychiatry 47: 213–219
First MB, Spitzer RL, Gibbon M, Williams J . (1996): Structured Clinical Interview for DSM-IV Axis I disorders (SCID), Clinician version: User's guide. Washington DC: American Psychiatric Press
Frey KA, Bohnen NI, Meyer P, Minoshima S, Wernette K, Koeppe RA, Kilbourn MR, Kuhl DE . (1998): Presynaptic monoaminergic vesicles in normal aging studied by (+)-[C-11]DTBZ PET. J Nucl Med 39S: 15
Frey KA, Holthoff VA, Koeppe RA, Jewett DM, Kilbourn MR, Kuhl DE . (1991): Parametric in vivo imaging of benzodiazepine receptor distribution in human brain. Ann Neurol 30: 663–672
Frey KA, Koeppe RA, Mulholland GK, Jewett D, Hichwa R, Ehrenkaufer RL, Carey JE, Wieland DM, Kuhl DE, Agranoff BW . (1992): In vivo muscarinic cholinergic receptor imaging in human brain with [11C]scopolamine and positron emission tomography. J Cereb Blood Flow Metab 12: 147–154
Gilman S, Koeppe RA, Adams KM, Junck L, Kluin KJ, Johnson-Greene D, Martorello S, Heumann M, Bandekar R . (1998): Decreased striatal monoaminergic terminals in severe chronic alcoholism demonstrated with (+)[11C]dihydrotetrabenazine and positron emission tomography. Ann Neurol 44: 326–333
Gjedde A, Reith J, Wong DF . (1996): In schizophrenia, some dopamine D2-like receptors are still elevated. Psychiatry Res 67: 159–162
Henry JP, Scherman D . (1989): Radioligands of the vesicular monoamine transporter and their use as markers of monoamine storage vesicles. Biochem Pharmacol 38: 2395–2404
Hietala J, Syvalahti E, Vuorio K, Nagren K, Lehikoinen P, Ruotsalainen U, Rakkolainen V, Lehtinen V, Wegelius U . (1994): Striatal D2 dopamine receptor characteristics in neuroleptic-naive schizophrenic patients studied with positron emission tomography. Arch Gen Psychiatry 51: 116–123
Hietala J, Syvalahti E, Vuorio K, Rakkolainen V, Bergman J, Haaparanta M, Solin O, Kuoppamaki M, Kirvela O, Ruotsalainen U . (1995): Presynaptic dopamine function in striatum of neuroleptic-naive schizophrenic patients. Lancet 346: 1130–1131
Hirai M, Kitamura N, Hashimoto T, Nakai T, Mita T, Shirakawa O, Yamadori T, Amano T, Noguchi KS, Tanaka C . (1988): [3H]GBR-12935 binding sites in human striatal membranes: Binding characteristics and changes in parkinsonians and schizophrenics. Jap J Pharmacol 47: 237–243
Hitri A, Casanova MF, Kleinman JE, Weinberger DR, Wyatt RJ . (1995): Age-related changes in [3H]GBR 12935 binding site density in the prefrontal cortex of controls and schizophrenics. Biol Psychiatry 37: 175–182
Ichinose H, Ohye T, Fujita K, Pantucek F, Lange K, Riederer P, Nagatsu T . (1994): Quantification of mRNA of tyrosine hydroxylase and aromatic L-amino acid decarboxylase in the substantia nigra in Parkinson's disease and schizophrenia. J Neural Transm Parkinsons Dis Dem Sect 8: 149–158
Jentsch JD, Taylor JR, Roth RH . (1998): Subchronic phencyclidine administration increases mesolimbic dopaminergic system responsivity and augments stress- and psychostimulant-induced hyperlocomotion. Neuropsychopharmacology 19: 105–113
Jernigan TL, Zisook S, Heaton RK, Moranville JT, Hesselink JR, Braff DL . (1991): Magnetic resonance imaging abnormalities in lenticular nuclei and cerebral cortex in schizophrenia. Arch Gen Psychiatry 48: 881–890
Jewett DM, Kilbourn MR, Lee LC . (1997): A simple synthesis of [11C]dihydrotetrabenazine (DTBZ). Nucl Med Biol 24: 197–199
Joyce JN, Lexow N, Bird E, Winokur A . (1988): Organization of dopamine D1 and D2 receptors in human striatum: Receptor autoradiographic studies in Huntington's disease and schizophrenia. Synapse 2: 546–557
Koeppe RA, Frey KA, Kuhl DE, Kilbourn MR . (1999): Assessment of extrastriatal vesicular monoamine transporter binding site density using stereoisomers of [11C]dihydrotetrabenazine. J Cereb Blood Flow Metab 19: 1376–1384
Koeppe RA, Frey KA, Kume A, Albin R, Kilbourn MR, Kuhl DE . (1997): Equilibrium versus compartmental analysis for assessment of the vesicular monoamine transporter using (+)-alpha-[11C]dihydrotetrabenazine (DTBZ) and positron emission tomography. J Cereb Blood Flow Metab 17: 919–931
Koeppe RA, Frey KA, Mulholland GK, Kilbourn MR, Buck A, Lee KS, Kuhl DE . (1994): [11C]tropanyl benzilate-binding to muscarinic cholinergic receptors: Methodology and kinetic modeling alternatives. J Cereb Blood Flow Metab 14: 85–99
Koeppe RA, Frey KA, Vander Borght T, Karlamangla A, Jewett DM, Lee LC, Kilbourn MR, Kuhl DE . (1996): Kinetic evaluation of [11C]dihydrotetrabenazine by dynamic PET: Measurement of vesicular monoamine transporter. J Cereb Blood Flow Metab 16: 1288–1299
Laruelle M, Abi DA, van Dyck CH, Gil R, D'Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB . (1996): Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci U S A 93: 9235–9240
Lauriello J, Hoff A, Wieneke MH, Blankfeld H, Faustman WO, Rosenbloom M, DeMent S, Sullivan EV, Lim KO, Pfefferbaum A . (1997): Similar extent of brain dysmorphology in severely ill women and men with schizophrenia. Am J Psychiatry 154: 819–825
Lim KO, Tew W, Kushner M, Chow K, Matsumoto B, DeLisi LE . (1996): Cortical gray matter volume deficit in patients with first-episode schizophrenia. Am J Psychiatry 153: 1548–1553
Lindstrom LH, Gefvert O, Hagberg G, Lundberg T, Bergstrom M, Hartvig P, Langstrom B . (1999): Increased dopamine synthesis rate in medial prefrontal cortex and striatum in schizophrenia indicated by L-(beta-11C) DOPA and PET. Biol Psychiatry 46: 681–688
McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA . (1998): Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: Evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci 18: 8417–8422
Minoshima S, Koeppe RA, Frey KA, Kuhl DE . (1994): Anatomic standardization: Linear scaling and nonlinear warping of functional brain images. J Nucl Med 35: 1528–1537
Mitchell IJ, Cooper AJ, Griffiths MR, Barber DJ . (1998): Phencyclidine and corticosteroids induce apoptosis of a subpopulation of striatal neurons: a neural substrate for psychosis? Neuroscience 84: 489–501
Naudon L, Leroux-Nicollet I, Costentin J . (1994): Short-term treatments with haloperidol or bromocriptine do not alter the density of the monoamine vesicular transporter in the substantia nigra. Neurosci Lett 173: 1–4
Nordstrom AL, Farde L, Eriksson L, Halldin C . (1995): No elevated D2 dopamine receptors in neuroleptic-naive schizophrenic patients revealed by positron emission tomography and [11C]NMSP. Psychiatry Res 61: 67–83
Overall JE, Gorham DR . (1962): Brief psychiatric rating scale. Psychol Rep 10: 799–812
Pearce RK, Seeman P, Jellinger K, Tourtellotte WW . (1990): Dopamine uptake sites and dopamine receptors in Parkinson's disease and schizophrenia. Eur Neurol 1: 9–14
Pilowsky LS, Costa DC, Ell PJ, Verhoeff NP, Murray RM, Kerwin RW . (1994): D2 dopamine receptor binding in the basal ganglia of antipsychotic- free schizophrenic patients. An 123I-IBZM single photon emission computerised tomography study. Br J Psychiatry 164: 16–26
Reith J, Benkelfat C, Sherwin A, Yasuhara Y, Kuwabara H, Andermann F, Bachneff S, Cumming P, Diksic M, Dyve SE, Etienne P, Evans AC, Lal S, Sherell M, Savard G, Wong DF, Chouinard G, Gjedde A . (1994): Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci U S A 91: 11651–11654
Seeman P . (1987): Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse 1: 133–152
Seeman P, Bzowej NH, Guan HC, Bergeron C, Becker LE, Reynolds GP, Bird ED, Riederer P, Jellinger K, Watanabe S, Tourtellotte WW . (1987): Human brain dopamine receptors in children and aging adults. Synapse 1: 399–404
Seiden LS, Fischman MW, Schuster CR . (1976): Long-term methamphetamine induced changes in brain catecholamines in tolerant rhesus monkeys. Drug Alcohol Depend 1: 215–219
Selemon LD, Rajkowska G, Goldman-Rakic PS . (1995): Abnormally high neuronal density in the schizophrenic cortex: A morphometric analysis of prefrontal area 9 and occipital area 17. Arch Gen Psychiatry 52: 805–818
Snyder SH . (1972): Catecholamines in the brain as mediators of amphetamine psychosis. Arch Gen Psychiatry 27: 169–179
Sonsalla PK, Nicklas WJ, Heikkila RE . (1989): Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science 243: 398–400
Talairach J, Tournoux P . (1988): A Co-Planar Stereotaxic Atlas of a Human Brain. Stuttgart: Thieme-Verlag
Vander Borght TM, Kilbourn M, Desmond T, Kuhl D, Frey K . (1995a): The vesicular monoamine transporter is not regulated by dopaminergic drug treatments. Eur J Pharmacol 294: 577–583
Vander Borght TM, Kilbourn MR, Koeppe RA, DaSilva JN, Carey JE, Kuhl DE, Frey KA . (1995b): In vivo imaging of the brain vesicular monoamine transporter. J Nucl Med 36: 2252–2260
Villemagne V, Yuan J, Wong DF, Dannals RF, Hatzidimitriou G, Mathews WB, Ravert HT, Musachio J, McCann UD, Ricaurte GA . (1998): Brain dopamine neurotoxicity in baboons treated with doses of methamphetamine comparable to those recreationally abused by humans: evidence from [11C]WIN-35,428 positron emission tomography studies and direct in vitro determinations. J Neurosci 18: 419–427
Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley SJ, Hitzemann R, Smith G, Fields SD, Gur R . (1996a): Dopamine transporters decrease with age. J Nucl Med 37: 554–559
Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, MacGregor RR, Schlyer DJ, Hitzemann R, Wolf AP . (1996b): Measuring age-related changes in dopamine D2 receptors with 11C-raclopride and 18F-N-methylspiroperidol. Psychiatry Res 67: 11–16
Wagner GC, Ricaurte GA, Johanson CE, Schuster CR, Seiden LS . (1980): Amphetamine induces depletion of dopamine and loss of dopamine uptake sites in caudate. Neurology 30: 547–550
Wise CD, Stein L . (1973): Dopamine-beta-hydroxylase deficits in the brains of schizophrenic patients. Science 181: 344–347
Wong DF, Wagner HJ, Tune LE, Dannals RF, Pearlson GD, Links JM, Tamminga CA, Broussolle EP, Ravert HT, Wilson AA, Toung JKT, Malat J, Williams JA, O'Tuama LA, Snyder SH, Kuhar MJ, Gjede A . (1986): Positron emission tomography reveals elevated D2 dopamine receptors in drug-naive schizophrenics. Science 234: 1558–1563
Young EA, Neff NH, Hadjiconstantinou M . (1993): Evidence for cyclic AMP-mediated increase of aromatic L-amino acid decarboxylase activity in the striatum and midbrain. J Neurochem 60: 2331–2333
Zipursky RB, Lim KO, Sullivan EV, Brown BW, Pfefferbaum A . (1992): Widespread cerebral gray matter volume deficits in schizophrenia. Arch Gen Psychiatry 49: 195–205
Acknowledgements
The authors whish to acknowledge the expert technical support of the PET center. Thanks are due to Laura Decker and Linda Ohl for their help in subject assessment. Financial support for this study came from the Michigan Mental Illness Research Association. SFT was supported by the National Institutes of Mental Health (K08 MH01258). All authors report no involvement presenting any possible conflicts of interest with the findings presented herein.
Author information
Authors and Affiliations
Additional information
This work was previously presented at 53rd annual meeting of the Society of Biological Psychiatry, Toronto, May 1998
Rights and permissions
About this article
Cite this article
Taylor, S., Koeppe, R., Tandon, R. et al. In Vivo Measurement of the Vesicular Monoamine Transporter in Schizophrenia. Neuropsychopharmacol 23, 667–675 (2000). https://doi.org/10.1016/S0893-133X(00)00165-2
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1016/S0893-133X(00)00165-2
Keywords
This article is cited by
-
Structural requirement of C11b chirality of tetrabenazine analogs as VMAT2 imaging ligands: synthesis and in vivo evaluation
Journal of Radioanalytical and Nuclear Chemistry (2017)
-
Presynaptic Dopaminergic Function: Implications for Understanding Treatment Response in Psychosis
CNS Drugs (2014)
-
Dopamine hypothesis of schizophrenia: Making sense of it all
Current Psychiatry Reports (2007)
-
PET Studies of Net Blood—Brain Clearance of FDOPA to Human Brain: Age-Dependent Decline of [18F]Fluorodopamine Storage Capacity
Journal of Cerebral Blood Flow & Metabolism (2005)
