
Abstract
Anorexia nervosa (AN) is a complex and multifactorial disorder occurring predominantly in women. Despite having the highest mortality among psychiatric conditions, it still lacks robust and effective treatment. Disorders such as AN are most likely syndromes with multiple genetic contributions, however, genome-wide studies have been underpowered to reveal associations with this uncommon illness. Here, we generated induced pluripotent stem cells (iPSCs) from adolescent females with AN and unaffected controls. These iPSCs were differentiated into neural cultures and subjected to extensive transcriptome analysis. Within a small cohort of patients who presented for treatment, we identified a novel gene that appears to contribute to AN pathophysiology, TACR1 (tachykinin 1 receptor). The participation of tachykinins in a variety of biological processes and their interactions with other neurotransmitters suggest novel mechanisms for how a disrupted tachykinin system might contribute to AN symptoms. Although TACR1 has been associated with psychiatric conditions, especially anxiety disorders, we believe this report is its first association with AN. Moreover, our human iPSC approach is a proof-of-concept that AN can be modeled in vitro with a full human genetic complement, and represents a new tool for understanding the elusive molecular and cellular mechanisms underlying the disease.
Similar content being viewed by others

Introduction
Anorexia nervosa (AN) is a multifactorial neurodevelopmental disorder of unknown etiology affecting around 1% of the population.1 With a stereotypic post-pubertal onset, this complex neuropsychiatric condition is primarily seen in female adolescents and young women between 15–19 years old.2, 3, 4 Anorexia presents with distorted body image and food restriction to the point of severe emaciation or death, likely resulting from a highly anxiogenic response to intake of palatable foods.5 Although exhibiting the highest mortality rates among psychiatric illnesses, with patients facing consequences such as high chronicity and morbidity, there are no treatments to reverse AN symptoms.6, 7, 8 Understanding the pathophysiology of AN has lagged behind other psychiatric disorders, consequently, eating disorders in general and AN in particular, have been viewed as non-biologically based problems of vanity, poor parenting or pertaining to specific groups of individuals.9 Several studies suggest that genes contribute significantly to neurobiological vulnerabilities, accounting for approximately 50–75% of the risk for AN.4, 10, 11 However, the identity of specific genes underlying the disease remains largely unexplored and the mechanisms are poorly understood. AN appears to aggregate in families and relatives of affected individuals seem to have an elevated risk of obsessive-compulsive personality disorder.12 It is also suggested that first degree relatives carry a relative risk of 11.3 compared to the general population,12, 13 with mothers showing increased rates of affective, substance use and anxiety disorders.14 In addition, various genetic studies have implicated a network of risk-conferring genes involved in serotonin and dopamine neurotransmission.15, 16, 17, 18, 19, 20 Functional neuroimaging studies corroborated these hypotheses19, 21, 22, 23, 24 by revealing a dysfunctional dopaminergic (DA) reward circuit in AN.
Despite progress in assigning circuits to neuropsychiatric disorders, disease-specific molecular and cellular phenotypic data for regions of interest is either limited, skewed by chronic illness in post-mortem samples or absent. This problem can now be addressed by reprogramming somatic cells into a pluripotent state by ectopic expression of specific genes.25 Such induced pluripotent stem cells (iPSCs) can be differentiated into neurons and have been generated for several complex neurological conditions.26, 27, 28, 29, 30, 31 Using this approach, we are able to test hypotheses and to evaluate whether the captured genome from human individuals afflicted with AN alters cellular phenotypes as predicted by mechanistic models of the disease. An AN-iPSC model may address human-specific effects and avoid some aspects of the well-known limitations of animal models such as the absence of a human genetic background.32
Here, we examined the expression profile of human AN-derived neurons after generating iPSCs from AN patients. We accessed not only targeted gene expression levels but we also performed whole transcriptome-based bioinformatics to reveal AN-risk conferring transcripts/genes that could be associated with disease initiation and progression. Our study represents a proof-of-concept that AN can be modeled in vitro. Although no predicted differences at neurotransmitters levels were observed, we identified a disruption of the tachykinin system that might contribute to AN pathophysiology before other phenotypes become prominent. While we believe that further validation using a larger cohort of patients is important, our work brings a novel technological advancement to the field of eating disorders. These findings could transform our ability to study how AN risk-conferring genetic variations perturb molecular pathways and cellular networks, highlighting potential approaches for new therapies.
Materials and methods
Participant ascertainment
We isolated and derived fibroblasts from seven females with AN, and four healthy female controls. The biopsied individuals, control CTL1 and patient AN1, are sisters. Fibroblast donations were solicited from patients who were receiving treatment at the outpatient clinic of the UCSD Eating Disorders Treatment and Research Center. For inclusion, patients had to be female and meet Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV)33 criteria for AN, except for the amenorrhea criterion. AN individuals exhibited a stereotypical, post-pubertal onset of weight loss from self-reducing food intake, fear of weight gain while underweight, with or without compensatory behaviors such as over-exercise and purging. In order to reduce a major source of heterogeneity, AN patients were selected for severity of symptoms and presence of serious medical and behavioral consequences of AN-related behaviors. This was feasible because all AN participants were recruited at a tertiary center for specialized, intensive eating disorders treatment, while receiving treatment under one of the authors. These recruitment characteristics ensured that the AN cohort not only met DSM-IV criteria but also met more stringent and homogeneous medical necessity criteria for severity of illness. AN individuals in our cohort had a relentless drive to lose weight that had actually resulted in weight loss. Treatment with their outpatient providers and efforts at home to restore weight was met with severe emotional dysregulation and/or medical abnormalities from worsening eating disordered behaviors. Furthermore, we were able to clearly assess these symptoms as the parents of the patients were seeking treatment for their adolescent daughters proximal to onset of their illness. AN individuals who over-exercised as the only compensatory method as well as others who used purging via self-induced vomiting, laxatives, among others, were included. Donations from healthy controls were solicited from siblings of AN patients along with unaffected individuals participating in other ongoing research studies. For inclusion, controls had to be female and should have never met an Axis I diagnosis per DSM-IV. Briefly, fibroblasts were obtained from skin punch biopsy performed by a Staff Dermatologist at the UCSD Outpatient Dermatology Clinic. Written informed consent was acquired from volunteers and their parents (when applicable), and all human research protocols were approved by the UCSD Institutional Review Board.
Generation of iPSCs
Skin fibroblasts from 8 individuals, that is, 4 AN patients and 4 controls, were reprogrammed into iPSCs. Cells were transduced with four retroviral constructs (OCT4, SOX2, KLF4 and c-MYC), as previously described.25, 30 Forming iPSCs were transferred to inactivated mouse embryonic fibroblast (MEF) feeders in human embryonic stem cell (hESC) medium (DMEM/F12 (Corning, Life Technologies, Carlsbad, CA, USA), 20% Knockout Serum Replacement (KSR; Life Technologies), 20 ng ml−1 basic fibroblast growth factor (bFGF; Life Technologies), 2 mm Glutamax (Gibco, Life Technologies), 1% non-essential aminoacids (NEAA; Sigma-Aldrich, St Louis, MO, USA), 0.1 mm β-mercaptoethanol (Sigma-Aldrich) and 1 mm of valproic acid (Sigma-Aldrich). hESC-like colonies, characterized by their compact morphology with a high nucleus-to-cytoplasm ratio, were manually dissected and passaged to pre-coated Matrigel (BD Biosciences, San Jose, CA, USA) plates in mTeSR1 medium (Stem Cell Technologies, Vancouver, BC, Canada). A minimum of 2 iPSC clones from each individual were selected for expansion.
Karyotyping
Standard G-banding karyotype from fibroblasts and iPSC clones was performed by the Stem Cell Core Facility at USC (Los Angeles, CA, USA), in collaboration with the Children’s Hospital Los Angeles.
Teratoma formation
Feeder-free fully grown iPSCs were dissociated with collagenase for 3–4 min at 37 °C and re-suspended in PBS and Matrigel (1:1). Next, 1–3 × 106 cells were injected subcutaneously in the lower hind leg and near the ankle of immune compromised mice. After 8 weeks, the tumor was dissected, fixed in 4% paraformaldehyde and paraffin embedded. For histological studies, sections of 5 μm thickness were stained with hematoxylin and eosin, and analyzed for the presence of the three different germ layer tissues. Protocols were approved by the UCSD Institutional Animal Care and Use Committee.
Embryoid body formation
Assessment of the tri-lineage differentiation capacity of iPSCs was performed in vitro through embryoid body (EB) formation. Briefly, iPSCs were mechanically dissociated and cultured in suspension for 14 days in non-adherent dishes to form EBs in DMEM-F12, 20% FBS. The presence of the three germ layers was investigated by gene expression (Supplementary Table S1).
Differentiation of iPSCs into cortical neurons
IPSC medium was changed to N2 medium comprising DMEM/F12 with l-Glutamine and 15 mm HEPES, 1 × N2 NeuroPlex (Gemini Bio-Products, West Sacramento, CA, USA), 1 μm dorsomorphin (R&D System, Minneapolis, MN, USA) and 10 μm SB431542 (Stemgent, Lexington, MA, USA) for 2 days. Next, cells were grown in suspension for 7 days in N2 medium. The formed EBs were gently dissociated and plated onto Matrigel-coated dishes using neural induction (NI) medium (DMEM/F12 with l-Glutamine and 15 mm HEPES, 0.5 × N2 NeuroPlex, 1 × Gem21 NeuroPlex (Gemini Bio-Products) and 20 ng ml−1 bFGF)). Rosettes that emerged were manually picked, dissociated with Accutase (Life Technologies) and re-plated onto 10 μg ml−1 Poly-l-ornithine and 5 μg ml−1 laminin-coated (from Sigma-Aldrich and Life Technologies, respectively) plates. Homogeneous populations of neural progenitor cells (NPC) were expanded using NI medium. The differentiation into neurons was performed upon bFGF withdrawal and addition of 5 μM ROCK inhibitor (Y-27632, Calbiochem, La Jolla, CA, USA). Cells were cultivated for 4 weeks with media changes every 3–4 days.
Immunocytochemistry
Cells were fixed with paraformaldehyde, permeabilized and blocked in 3% bovine serum albumin and 0.1% Triton X-100. Primary antibodies were incubated overnight at 4 °C; secondary antibodies were incubated for 1 h at room temperature. Nuclei were stained with DAPI solution (1 μg ml−1). Slides were mounted using Prolong Gold antifade reagent (Life Technologies). Images were captured using a Zeiss microscope (Carl Zeiss, Jena, Germany). For antibodies specifications, refer to Supplementary Table S2.
Gene expression analysis
Total RNA was reverse transcribed with QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA, USA). To investigate the expression of pluripotency and the three germ layer genes, cDNA from iPSCs and EBs was amplified by PCR. qRT-PCR was also performed for selected genes using specific primers and iQ SYBR Green supermix (Bio-Rad Laboratories, Irvine, CA, USA). The expression of each target gene and the reference gene (B2M, beta-2-microglobulin and/or GAPDH, Glyceraldehyde-3-phosphate dehydrogenase) was measured in technical triplicates for each reaction. The relative expression quantification was normalized using the 2(−ΔΔCt) method. The AN and control groups were compared by Student’s t-test. For primer sequences, refer to Supplementary Table S1. The Human Neurotransmitter Receptors RT2 Profiler PCR Array (#PAHS-060Z; SABiosciences, Qiagen, Valencia, CA, USA) was used for the quantitative gene expression analysis of neuronal cultures, as suggested by the manufacturer.
Western blotting
Twenty micrograms of total protein extracts were separated in Bolt 4–12% Bis-Tris Plus Gel (Life Technologies) and transferred onto a nitrocellulose membrane using Thermo Fisher’s iBlot2 dry blotting system. After blocking (Rockland Immunochemicals, VWR International, Arlington Heights, IL, USA), membranes were incubated with primary antibodies overnight at 4 °C and then, with secondary antibodies for 1 h at room temperature. Proteins were detected using Odyssey CLx infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Transcriptome analysis
RNA-sequencing (RNA-seq) analysis was performed in 10 μg of total RNA using Illumina Hiseq-2000 (Illumina, San Diego, CA, USA). Raw sequenced RNA-seq libraries were filtered for high-quality reads based on read average quality, and per position nucleotide detection using NGS QC Toolkit software.34 High-quality reads were mapped to the human reference genome (Hg19) using Star aligner,35 producing compressed binary BAM files. Next, binary BAM files were subjected to HTSeq software package36 to account for absolute number of mapped reads per annotated transcript (Ensembl GRCh37 annotation) in Hg19, generating a count data matrix. This matrix was then normalized by a read counting approach followed by a negative binomial distribution and Fischer’s exact statistical test for differential expression (DE) analysis using the R Bioconductor package DESeq.37 For statistical significance calculation between samples, false discovery rate based on Benjamini & Hochberg method38 was applied over the DE genes, with DE transcripts having P-value<0.05. Count data was used to calculate Euclidian distance between each pair of samples, including biological replicates, which were clustered based on shorter distances. In addition, count data matrix was also used to create a heatmap together with a hierarchical clustering-based dendrogram, and a two-component-based Principal Component Analysis (PCA), to show relative relationship between sequenced AN and control samples in a 2D coordinate space. For molecular network and functional pathway annotation robustness, we considered in the analysis only those genes common to both annotation databanks, ENSEMBL (GRCh38.82) and HUGO Gene Nomenclature Committee (HGNC). There are 35 644 genes registered in the HGNC database and 60 619 genes in the ENSEMBL gene annotation database, with 35 588 genes commonly identified in both databanks.
Gene regulatory networks and functional pathways analyses
The protein–protein interaction network (PIN) was constructed by using the protein-coding differentially expressed genes (DEGs) and their direct neighbors (or first-degree interactors). The PIN data were retrieved from the Biogrid database (v 3.4.127).39 The statistical analysis of network properties and structure was carried out using Python programming language and igraph package (igraph.sourceforge.net). Network visualization and annotation were performed using Cytoscape40 (http://cytoscape.org/). ClueGO41 was used to find functional pathways enriched in AN-PIN based on functional annotation databases such as Gene Ontology42 (GO; http://geneontology.org/) and Reactome43 (http://www.reactome.org). For statistical analysis of PINs, we used a computational method published elsewhere44 to measure two structural properties: average shortest path length and density of the network. We tested whether the distribution of those structural properties was similar between AN-PIN and random PIN. The non-parametric Kolmogorov-Smirnov test as implemented in R (www.r-project.org) was used to test whether the structural properties of AN-PIN were similar to random PINs (null hypothesis). The transcriptional profiles of specific genes in the developing human brain were investigated using available RNA sequencing data from BrainSpan database (http://www.brainspan.org).45 These data were generated across 13 developmental stages in 8–16 brain structures.
Results
Reprogramming somatic cells from AN patients and controls into iPSCs
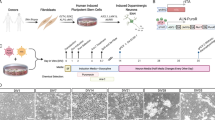
All AN patients included in this study had met the DSM-IV criteria for AN; control individuals (CTL) had never met any Axis I diagnosis. AN1 and CTL1 are siblings. The phenotypic characterization of AN subjects is presented in Figure 1a. A schematic view of our protocol for iPSC reprogramming and differentiation is shown in Figure 1b, and the cell types used in each experiment is detailed in Supplementary Table S3. Two iPSC clones from 4 AN patients and 4 controls, in a total of 16 cell lines, were mechanically expanded for at least 10 passages and tested for the expression of the pluripotency markers OCT4 (also known as POU5F1), TRA-1-60, NANOG and LIN28 (Figure 1c; Supplementary Figure 1a, b). Apart from sample AN2, which exhibited an extra structurally abnormal chromosome (ESAC) that was also present in the original fibroblasts, all iPSC clones derived had maintained a normal karyotype when analyzed by G-banding (Figure 1d; Supplementary Figure S1c).

Generation and characterization of iPSCs from AN patients. (a) Patient profile summary. (b) Schematic view and representative images showing the morphological changes observed during the primary fibroblast cell reprogramming, neural induction and differentiation processes. Scale bars represent 200 μm. (c) Representative immunofluorescence images illustrating the expression of pluripotency markers in the generated iPSCs, including OCT4, NANOG, TRA-1–60 and LIN28. Scale bar represents 100 μm. (d) Representative images of G-banding karyotype analysis from cell chromosomes demonstrating the genetic stability of iPSCs; no karyotypic abnormalities were induced by the reprogramming process. (e) Representative images of hematoxylin and eosin staining of teratomas derived from iPSCs showing tissues from the three germ layers. Scale bar represents 100 μm. (f) Expression of pluripotency and three germ layer markers in iPSCs and EBs, respectively, assessed by RT-PCR (OCT4, NANOG and LIN28—pluripotency; AFP—endoderm; MSX1—mesoderm; PAX6—ectorderm). The H9-hESC was used as a control for pluripotency and differentiation capability; B2M was used as reference gene. (g) Cluster analysis showing correlation coefficients of RNA-seq transcripts from iPSCs and hESC, and a distinguished gene expression profile from primary fibroblast cells (FIBRO). A panel of human pluripotency-related genes (isoform level; Supplementary Table S3) was considered. (h) Heatmap and hierarchical clustering-based dendrogram of hESC, iPSCs and fibroblasts for AN and control samples. Considering the entire cellular transcriptome expression profile of evaluated cells, two subgroups were identified: iPSCs, with a molecular signature similar to that exhibited by hESCs, and fibroblasts with a completely different expression profile. In g and h, colors indicate the range of each gene’s expression, with least expression shown in red and highest expression shown in green. AN, anorexia nervosa; anxiety, patient showed/treated for anxiety; CTL, control (unaffected individual); famhx-ED, first, second or third degree relative with a history of an eating disorder; fi, fibroblast; iPSC, induced pluripotent stem cell; MDD, major depressive disorder; neu, neurons; NPC, neural progenitor cell; OCD, obsessive compulsive disorder; psychotropic, patient was prescribed at least one psychotropic medication.
The ability of clones to differentiate into three germ layers was evaluated in vivo and in vitro. Teratomas showed derivatives from all three embryonic germ layers (ecto-, meso- and endoderm), confirming that the iPSCs were pluripotent and able to differentiate into complex tissues in vivo (Figure 1e). The EBs generated in suspension were tested by RT-PCR and we observed that the expression of endogenous pluripotency markers characteristic for hESC were exhibited in all reprogrammed iPSCs (OCT4, NANOG and LIN28), while genes related with the three germ layers were only identified in the EBs (AFP, α-fetoprotein—endoderm; MSX1, Msh homeobox 1—mesoderm; PAX6, paired box homeotic gene 6—ectoderm; Figure 1f). In addition, considering a panel of 8 different genes (and their isoforms) associated with pluripotency, we compared the iPSCs and their fibroblasts counterparts used for the reprogramming process with a hESC line (H9; Supplementary Table S4). The high throughput RNA-seq and bioinformatics data analysis of these cells showed that the iPSC clones were indistinguishable from hESC lines and were also very similar to each other, but distinct from primary fibroblast cells (Figure 1g). Next, by investigating the global transcriptome expression signatures of these cell types we identified two subgroups, iPSC/hESC and fibroblasts, which have completely distinct expression profiles and yet a high degree of similarity within each subgroup when examined for pluripotency-related gene markers (Figure 1h). Together, these data show that the generated iPSC lines successfully re-established pluripotency at the molecular and cellular levels.
Differentiation of AN and control iPSCs into neurons
For all 16 iPSC clones used in this study, dissociated rosettes formed a homogenous population of NPCs after a few passages, that is positive for early precursor markers such as NESTIN, MUSASHI1 and SOX2 (Figure 2a, Supplementary Figure S1d–f). Next, we compared the global molecular signatures from iPSC and neuronal cultures after 4 weeks of differentiation. Two different gene expression profiles were observed, confirming that we were able to efficiently differentiate the generated pluripotent cells into neurons (Figure 2b). The comparison of iPSC, NPC and neurons according to their levels of expression of OCT4, NESTIN and MAP2 clearly showed that OCT4 was highly expressed in iPSCs but not in the other cell types, while in progenitor cells and neurons NESTIN and MAP2 were preferentially upregulated, respectively (Figure 2c). Using the described protocol, NPC differentiation gave rise to a mature population of cortical human neurons expressing MAP2, GFAP, NEUN and CTIP2 (also known as BCL11B; cortical layer V; Figures 2d and e; Supplementary Figure S1g). Moreover, in these differentiated cultures we also detected the presence of both excitatory glutamatergic pyramidal-shaped neurons (VGLUT1 and SYN1) and inhibitory GABAergic interneurons (GAD65-67 and GABA) along with LMX1A and FOXA2 positive cells, which are selectively expressed in progenitors committed to the generation of midbrain dopaminergic neurons (Figure 2d and e; Supplementary Figure S1g-i). Western blot analysis corroborated the immunostaining results and confirmed that the overall composition of neuronal cultures does not change between controls and AN patients (Figures 2f and g; Supplementary Figure S2a, c).

Derivation of neural progenitor cells and neurons from AN-iPSCs. (a) Representative images showing that the neural progenitor stage-specific markers NESTIN and SOX2 are expressed by AN iPSC-derived NPCs. Scale bar represents 100 μm. (b) Cluster analysis showing that after 4 weeks of differentiation neurons (NE) exhibit a molecular signature distinct from that of their iPSC counterparts. Colors indicate the range of each gene’s expression, with least expression shown in red and highest expression shown in green. (c) Gene expression changes observed during the differentiation process measured by qRT-PCR using stage-specific markers for iPSCs (OCT4), NPCs (NESTIN) and neurons (MAP2). The expression levels of each gene were quantified, normalized to B2M (reference gene), and the results are presented as mean±s.e.m. (n⩾8 for each group). (d) Representative immunofluorescence images of cells after neuronal differentiation. IPSC-derived neural cultures express neuronal (MAP2, NEUN) and glial (GFAP) markers, together with specific cortical proteins (CTIP2). Excitatory (VGLUT1 and SYN1) and inhibitory (GAD65-67 and GABA) neuronal proteins are also observed in the generated neural population. The presence of LMX1A and FOXA2 among the neuronal cells, although low, is an evidence of the dopaminergic neuronal fate. Scale bar represents 100 μm. (e) Quantification of the percentage of MAP2+ (neuron), VGLUT1+ (glutamatergic), GABA+ (GABAergic) and GFAP+ (glia) labeled cells is presented as mean±s.e.m. (n⩾8 for each group). (f) Representative western blotting of control and AN-derived neural proteins that were lysed and immunoblot for neuronal (TUJ1, VGLUT1, GAD65-67, PSD95, SYN1 and TH) and glial (GFAP) markers, along with the dopamine transporter (DAT); β-ACTIN was used as housekeeping control (reference). (g) Quantification of proteins in AN and control neural cultures assessed by Western blot analysis; β-ACTIN was used for normalization (n=8 for each group). No differences were observed between control and affected samples (P<0.05, Student’s t-test). AN, anorexia nervosa; iPSC, induced pluripotent stem cell; NPC, neural progenitor cells.
Gene expression profile of iPSC-derived neurons
After 4 weeks of differentiation, the gene expression profile of AN neurons and controls was investigated. A total of 24 944 genes were found to be transcribed in the iPSC-derived neurons from AN patients and healthy individuals; 361 DEGs (156 upregulated and 205 down-regulated) were identified in AN compared to controls. The DEGs are annotated as four locus groups: 248 protein-coding genes, 50 non-coding RNAs, 57 pseudogenes and 6 others (Supplementary Table S5). The global molecular signature obtained after RNA-seq showed that both neuronal populations are very similar (Figure 3a). When we narrowed down this analysis considering only a panel of genes related to neural development and differentiation, no significant differences were observed (Figure 3b; Supplementary Table S6) suggesting that noticeable developmental anomalies might not be present in AN patients’ brain. After considering genes differentially expressed between affected and unaffected subjects with a statistical significance of 95% (fold change⩾2, P<0.05, 545 genes; Supplementary Table S7), we increased the stringency of our analysis by including only targets with 99% significance (fold-change⩾2, P<0.01, 110 genes; Supplementary Table S8) and found that AN samples cluster together in a subgroup different from that of control individuals (Figure 3c). Interestingly, when 13 candidate genes were selected based on their altered expression in AN with fold-change variation of 2 or more, the clustering algorithm produced subgroups with affected and control samples that remained distinct (Figure 3d). We validated the RNA-seq results for these 13 genes by qRT-PCR and confirmed the same expression patterns observed in the transcriptome analysis (Figure 3e), suggesting that such genes might contribute to the anorexic phenotype.

Transcriptional analysis of AN iPSC-derived neuronal cultures. (a) Heatmap and the hierarchical clustering-based dendrogram of samples after 4 weeks of differentiation. No significant differences are observed between iPSC-derived neurons (NE) from AN patients and controls. (b) Heatmap and the hierarchical clustering-based dendrogram displaying the transcriptional pattern of genes associated with neural development and differentiation in AN and control neural cultures (list of genes at Supplementary Table S5). (c) Heatmap and hierarchical clustering-based dendrogram of genes with minimum fold-change variation of 2 and false discovery rate (FDR)-adjusted P-value<0.01 between AN and control neurons. (d) Neuronal specific cell type-based clustering analysis of AN and control samples using 13 selected candidate genes most differentially expressed between the two subgroups (P<0.01). In all heatmaps (a–d), colors indicate the range of each gene’s expression, with least expression shown in red and highest expression shown in green. (e) Validation of 13 selected candidate genes from the RNA-seq analysis by qRT-PCR. Downregulated (red histogram) or upregulated (green histogram) genes differentially expressed between AN and control neurons. Independent neuronal cultures generated from the same clones used in the RNA-seq analysis were used in this validation; GAPDH was used as reference gene. Error bars are represented by standard deviation. (f) Topological structure of AN-PIN and random-PIN showing that AN-PIN is denser and with smaller shortest path than random-PIN (Kolmogorov–Smirnov test, P<0.01). (g) Enriched GO functional pathways terms found in neurons derived from AN patients (the complete list is found in the Supplementary Table S8). (h) Volcano plot of PCR array analysis for human neurotransmitter receptors. Plot illustrates that although control and AN-derived neurons do not show significant differences in expression for estrogen receptors and dopamine/serotonin neurotransmitter systems constituents, the TACR1 gene is upregulated in AN neurons (2.0-fold differential expression between the groups at P<0.05; Student’s t-test). (i) Upper panel: representative western blotting of control and AN-derived neural proteins that were lysed and immunoblot for TACR1; β-ACTIN was used as reference. Bottom panel: quantification of TACR1 protein in AN and control neural cultures assessed by Western blot analysis. Increased levels of TACR1 were observed in AN (P<0.05, Student’s t-test). Proteins were detected using Odyssey CLx infrared imaging system. (j) BrainSpan analysis of the TACR1 gene in brain regions from striatal networks. RNA-seq RPKM (reads per kilobase per million) identified during the different stages of human brain development. ACC, anterior cingulate cortex; AN, anorexia nervosa; iPSC, induced pluripotent stem cell; MNT, mediodorsal nucleus of thalamus; OFC, orbital frontal cortex; STR, striatum; VPC, ventrolateral prefrontal cortex.
To identify functional pathways and molecular interactions associated with DEGs in AN neurons, we first built a protein-protein interaction network (PIN) using the 248 protein-coding DEGs (P<0.01; Supplementary Table S5) and their direct neighbors (first-degree interactors). Approximately 68% of DEGs (168) were found in the protein-protein interaction database and the AN-PIN is comprised of 1492 proteins and 2254 interactions. The topological structure of AN-PIN is different from PIN built out of randomly selected genes indicating that DEGs in AN have higher connectivity than expected by chance (Kolmogorov-Smirnov test D=0.81, P<0.01), and smaller average shortest path (Kolmogorov–Smirnov test D=0.41, P<0.01; Figure 3f). These data suggest that proteins encoded by co-expressed genes in AN subjects are significantly more inter-connected and are likely to be associated with closely related molecular pathways including TACR1 (tachykinin receptor 1) signaling, regulation of synaptic transmission through the cholinergic system and response to estrogen, among others (Fisher’s exact test with Bonferroni correction P<0.01; Figure 3g; Supplementary Table S9).
Several neurotransmitters and peptides, or their metabolites, can potentially contribute to the AN phenotype, including weight loss, feeding regulation and the reward system.15, 22, 46, 47 Then, based on the enriched pathways revealed by the DEGs of our sample cohort, we decided to investigate the neurotransmitter profile of AN-derived neuronal cultures using a PCR array platform. This approach was complemented by the addition of specific targets pertaining to the dopaminergic pathway, estrogen receptors and tachykinin signaling (Supplementary Table S1). Notably, both control and AN neurons are very similar except by the expression of the TACR1 gene, also known as NK1R (neurokinin 1 receptor) or SPR (substance P receptor), which was significantly upregulated in AN (Figure 3h). In confirmatory studies, the TACR1 gene was found upregulated after RNA-seq analysis with a significance of 95% (Supplementary Table S7), while the TAC1 (tachykinin precursor 1) gene had a reduced expression of almost two fold-change (Supplementary Table S7 and S8) in AN neuronal samples. The protein levels of TACR1 were also increased in AN neurons compared to controls (Student’s t-test, P<0.05; Figure 3i; Supplementary Figure S2a, b). Furthermore, the BrainSpan analysis of the TACR1 gene within brain regions associated with striatal networks showed its expression peaks during adolescence, the stage of human development in which AN has its onset (Figure 3j). Although TACR1 has previously been associated with psychiatric disorders, we believe this is the first report of its contribution to AN.
Discussion
AN is a severe psychiatric disorder that still lacks effective treatments.7, 8 Several studies have been conducted aiming to identify the genetic basis of AN. Genome-wide association approaches (GWAS) have emerged as a promising tool for genetic screening. Even though single-nucleotide polymorphisms and/or copy-number variations have been identified, these findings require further analysis in independent cohorts and no study was powerful enough to robustly link specific genes with AN.10, 48, 49, 50, 51, 52 The difficulty in validating potential candidates likely arises from the need for large sample populations, which is particularly difficult for uncommon disorders like AN, that has an incidence far <1% of adolescents. Therefore, the genetic underlying AN susceptibility remains largely unknown. The literature in AN is mostly derived from DNA-based assays performed in blood samples or animal models. In this context, the ability to perform a transcriptome analysis to uncover genes implicated in AN pathophysiology using human neural cells could provide a major advantage toward understanding the etiology of the disorder.
Following the reprogramming of skin fibroblasts into iPSCs, we generated neural cultures that allowed us to investigate the gene expression profile of neurons derived from AN patients. The global molecular signature of neurons after RNA-seq analysis showed that AN and control cultures are, overall, very similar to each other. However, two subgroups were identified via a high-stringency analysis using differentially expressed genes with a statistical significance of 99% between affected and unaffected individuals. Among the misregulated genes found, CTGF (connective tissue growth factor), which is critical for normal ovarian follicle development and ovulation,53 was downregulated in AN samples. This result is congruent with the amenorrhea symptom typically observed in patients.54 The misregulation of CTGF in our cohort, despite not using amenorrhea as a diagnostic criterion, might serve as preliminary support for omission of this criterion. The lower expression of TDRD10 (tudor domain containing 10) could also contribute to AN pathophysiology, since the TDRD family of proteins is known to be relevant for gametogenesis55 and anorexics became at least temporarily infertile.56 Interestingly, the FABP12 (Fatty-acid Binding Protein 12) gene, which is related to lipid metabolism as other FABP members,57 was downregulated in AN-neural cultures, accompanied by high expression levels of TSHR. Since TSHR is detected in preadipocytes and adipocytes,58, 59 and was shown to participate in adipogenesis,60 disruption in this gene was already associated with body weight and energy consumption problems.61 Chen et al.62 demonstrated that increased TSHR inhibits FASN (fatty acid synthase) expression or energy storage in mature adipocytes, showing that impairments in TSH/TSHR signaling could participate in the development of obesity. In the same way, the DGKG (Diacylglycerol Kinase Gamma) gene, that had been associated with chronic stress63 and obesity,64 was found upregulated in AN-neurons. These findings suggest that misregulation of several genes, primarily unrelated, could contribute to the genesis and/or symptomatology of this multifactorial disease in a synergistic manner.
Despite the limitation of GWAS in finding statistical significance for risk-conferring genes in AN, a variety of studies have identified the serotonin and dopamine systems as major players in the pathophysiology of the disease.20, 21, 22, 65, 66, 67 However, the literature remains unsettled regarding these hypotheses.68, 69 Since mood and motor activity, food intake, decision-making abilities, reinforcement and reward are features directly affected by disruptions in the serotonin and/or dopamine pathways, we decided to investigate not only these systems but a broader neurotransmitter expression profile in neurons derived from AN patients. No substantial expression changes were found either among serotonin- nor among dopamine-related genes in our culture system. Notably, the expression of TACR1 was significantly upregulated in AN. This G-protein coupled receptor (GPCR) is distributed in many areas of the human brain including the frontal cortex,70 the amygdala and striatum,71 and has high affinity for Substance P (SP), the most abundant tachykinin (or neurokinin) that participates in the regulation of a variety of biological functions.72, 73
Several studies have reported the contribution of TACR1 to affective, anxiety and motivated disorders such as addictive behavior,74 bipolar disorder,75, 76, 77 attention deficit hyperactivity disorder,75, 78 depression,79, 80 fear81, 82 and anxiety.73, 81, 83, 84, 85 AN presents as a motivated disorder with anxiety3 induced by palatable food and is frequently comorbid with mood symptoms,3 especially while the patient is underweight. While TACR1 has been implicated in a variety of animal models that recapitulate core phenotypes of AN such as heightened anxiety and hyperactivity,78, 86, 87 absence of TACR1 also increases risk for high body mass index in a sex-dependent manner in mice.88 Conversely, increased TACR1 expression in human females with AN presumably increases risk for low body mass index. Among its diverse biological roles, TACR1 antagonists have been developed to treat emesis and reduction in food intake in those receiving cisplatin chemotherapy.89 Mechanistically, TACR1 and SP have been shown to interact with the monoaminergic system70, 83, 90, 91 and to modulate the reward pathway.92, 93 Notably, the Brainspan atlas show TACR1 expression peaking during adolescence in the striatum, a brain region implicated in impaired reward processing in AN. The onset of AN is during adolescence and brain imaging studies reveal structural and functional abnormalities in striatal networks.94 The pharmacology of reversing phenotypes in TACR1 models and use of dopamine antagonists in the treatment of AN share a common mechanism of targeting the dopaminergic system. Interestingly, a study in Caenorhabditis elegans showed that a tachykinin neuroendocrine signalling mediates body fat loss, by connecting the neural serotonin circuit to its metabolic actions in the intestine.95 Thus, many aspects of the disease onset and symptoms could be associated with misregulated tackykinins, including the serotonin and dopamine imbalances78 observed in AN clinical studies,19, 21, 22 especially due to the overlap and functional interaction of monoamines and SP in the brain.92, 96, 97, 98
In summary, we have developed a new human cellular model of AN and performed a complete and systematic transcriptional analysis of human AN-derived neurons. Despite using a small cohort, our study identified TACR1, a member of the tachykinin family that has previously been associated with other psychiatric disorders, dysregulated in AN. This gene arises as an unexplored target that potentially contributes to the pathophysiology of AN and that might be compromised in patients before other neurotransmitter systems are disrupted. Nonetheless, it is noteworthy that while our approach represents an innovative tool for understanding the molecular and genetic mechanisms contributing to AN, there is a caveat since the reprogramming process can alter the epigenetic memory of cells. If such alterations participate in the disease, they could be underestimated here and should be addressed in future studies. Improvements in the efficacy of current treatments and development of new therapies have been hampered by a lack of knowledge of the neurobiology of AN. Then, identifying novel molecular pathways altered in this complex disorder will provide an opportunity for new diagnostic strategies and treatments. Our study represents a proof-of-concept that AN can be modeled in vitro. Although our findings should be confirmed in a broader cohort of patients in the future, we believe that some features attributed to serotonin and/or dopamine pathways could be a consequence of a mis-regulated tachykinin system, ultimately affecting mood, food intake, lipid metabolism, anxiety and reward behaviors in AN individuals.
References
Smink FRE, van Hoeken D, Hoek HW . Epidemiology of eating disorders: incidence, prevalence and mortality rates. Curr Psychiatry Rep 2012; 14: 406–414.
Clarke TK, Weiss aRD, Berrettini WH . The genetics of anorexia nervosa. Clin Pharmacol Ther 2012; 91: 181–188.
Guarda AS, Schreyer CC, Boersma GJ, Tamashiro KL, Moran TH . Anorexia nervosa as a motivated behavior: Relevance of anxiety, stress, fear and learning. Physiol Behav 2015; 152: 466–472.
Bulik CM, Sullivan PF, Tozzi F, Furberg H, Lichtenstein P, Pedersen NL . Prevalence, heritability, and prospective risk factors for anorexia nervosa. Arch Gen Psychiatry 2006; 63: 305–312.
Sharan P, Sundar AS . Eating disorders in women. Indian J Psychiatry 2015; 57: S286–S295.
Arcelus J, Mitchell AJ, Wales J, Nielsen S . Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch Gen Psychiatry 2011; 68: 724–731.
Bulik CM, Berkman ND, Brownley KA, Sedway JA, Lohr KN . Anorexia nervosa treatment: a systematic review of randomized controlled trials. Int J Eat Disord 2007; 40: 310–320.
Watson HJ, Bulik CM . Update on the treatment of anorexia nervosa: review of clinical trials, practice guidelines and emerging interventions. Psychol Med 2013; 43: 2477–2500.
O’Hara SK, Smith KC . Presentation of eating disorders in the news media: What are the implications for patient diagnosis and treatment? Patient Educ Couns 2007; 68: 43–51.
Wang K, Zhang H, Bloss CS, Duvvuri V, Kaye W, Schork NJ et al. A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Mol Psychiatry 2011; 16: 949–959.
Brandys MK, de Kovel CG, Kas MJ, van Elburg AA, Adan RA . Overview of genetic research in anorexia nervosa: the past, the present and the future. Int J Eat Disord 2015; 48: 814–825.
Lilenfeld LR, Kaye WH, Greeno CG, Merikangas KR, Plotnicov K, Pollice C et al. A controlled family study of anorexia nervosa and bulimia nervosa: psychiatric disorders in first-degree relatives and effects of proband comorbidity. Arch Gen Psychiatry 1998; 55: 603–610.
Strober M . Controlled family study of anorexia nervosa and bulimia nervosa: evidence of shared liability and transmission of partial syndromes. Am J Psychiatry 2000; 157: 393–401.
Rost S, Kappel V, Salbach H, Schneider N, Pfeiffer E, Lehmkuhl U et al. Psychiatric disorders and personality styles in mothers of female adolescent patients with eating disorders. Z Kinder Jugendpsychiatr Psychother 2016; 1–9 (e-pub ahead of print).
Kaye WH, Frank GK, Bailer UF, Henry SE . Neurobiology of anorexia nervosa: Clinical implications of alterations of the function of serotonin and other neuronal systems. Int J Eat Disord 2005; 37: S15–S19.
Rask-Andersen M, Olszewski PK, Levine AS, Schiöth HB . Molecular mechanisms underlying anorexia nervosa: focus on human gene association studies and systems controlling food intake. Brain Res Rev 2010; 62: 147–164.
Kaye WH, Gwirtsman HE, George DT, Jimerson DC, Ebert MH . CSF 5-HIAA concentrations in anorexia nervosa: reduced values in underweight subjects normalize after weight gain. Biol Psychiatry 1988; 23: 102–105.
Phillipou A, Rossell SL, Castle DJ . The neurobiology of anorexia nervosa: a systematic review. Aust N Z J Psychiatry 2014; 48: 128–152.
Kaye WH, Frank GK, McConaha C . Altered dopamine activity after recovery from restricting-type anorexia nervosa. Neuropsychopharmacology 1999; 21: 503–506.
Gervasini G, Gordillo I, García-Herráiz A, Flores I, Jiménez M, Monge M et al. Influence of dopamine polymorphisms on the risk for anorexia nervosa and associated psychopathological features. J Clin Psychopharmacol 2013; 33: 551–555.
Frank GK, Kaye WH, Meltzer CC, Price JC, Greer P, McConaha C et al. Reduced 5-HT2A receptor binding after recovery from anorexia nervosa. Biol Psychiatry 2002; 52: 896–906.
Frank GK, Bailer UF, Henry SE, Drevets W, Meltzer CC, Price JC et al. Increased dopamine D2/D3 receptor binding after recovery from anorexia nervosa measured by positron emission tomography and [11c]raclopride. Biol Psychiatry 2005; 58: 908–912.
Wagner A, Aizenstein H, Venkatraman VK, Fudge J, May JC, Mazurkewicz L et al. Altered reward processing in women recovered from anorexia nervosa. Am J Psychiatry 2007; 164: 1842–1849.
Kaye WH, Gwirtsman HE, George DT, Ebert MH . Altered serotonin activity in anorexia nervosa after long-term weight restoration. Does elevated cerebrospinal fluid 5-hydroxyindoleacetic acid level correlate with rigid and obsessive behavior? Arch Gen Psychiatry 1991; 48: 556–562.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872.
Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010; 143: 527–539.
Mitne-Neto M, Machado-Costa M, Marchetto MCN, Bengtson MH, Joazeiro CA, Tsuda H et al. Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum Mol Genet 2011; 20: 3642–3652.
Andrade LN, de S, Nathanson JL, Yeo GW, Menck CFM, Muotri AR . Evidence for premature aging due to oxidative stress in iPSCs from Cockayne syndrome. Hum Mol Genet 2012; 21: 3825–3834.
Nageshappa S, Carromeu C, Trujillo CA, Mesci P, Espuny-Camacho I, Pasciuto E et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol Psychiatry 2015; 21: 178–188.
Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry 2015; 20: 1350–1365.
Chailangkarn T, Trujillo CA, Freitas BC, Hrvoj-Mihic B, Herai RH, Yu DX et al. A human neurodevelopmental model for Williams syndrome. Nature 2016; 536: 338–343.
Gutierrez E . A rat in the labyrinth of anorexia nervosa: contributions of the activity-based anorexia rodent model to the understanding of anorexia nervosa. Int J Eat Disord 2013; 46: 289–301.
Association AP Diagnostic and Statistical Manual of Mental Disorders, 4th edn. American Psychiatric Association: Washington, DC, 1994.
Patel RK, Jain M . NGC QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 2012; 7: e30619.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21.
Anders S, Pyl PT, Huber W . HTSeq - a Python framework to work with high-throughput sequencing data. Bioinformatics 2014; 31: 166–169.
Anders S, Huber W . Differential expression analysis for sequence count data. Genome Biol 2010; 11: R106.
Benjamini Y, Hochberg Y . Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 1995; 57: 289–300.
Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, Tyers M . BioGRID: a general repository for interaction datasets. Nucleic Acids Res 2006; 34: D535–D539.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13: 2498–2504.
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009; 25: 1091–1093.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25: 25–29.
Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G et al. The Reactome pathway knowledgebase. Nucleic Acids Res 2014; 42: D472–D477.
Cristino AS, Williams SM, Hawi Z, An J-Y, Bellgrove MA, Schwartz CE et al. Neurodevelopmental and neuropsychiatric disorders represent an interconnected molecular system. Mol Psychiatry 2014; 19: 294–301.
Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A et al. Transcriptional landscape of the prenatal human brain. Nature 2014; 508: 199–206.
Kaye WH, Ebert MH, Raleigh M, Lake R . Abnormalities in CNS monoamine metabolism in anorexia nervosa. Arch Gen Psychiatry 1984; 41: 350–355.
Kaye W . Neurobiology of anorexia and bulimia nervosa. Physiol Behav 2008; 94: 121–135.
Boraska V, Davis OSP, Cherkas LF, Helder SG, Harris J, Krug I et al. Genome-wide association analysis of eating disorder-related symptoms, behaviors, and personality traits. Am J Med Genet B Neuropsychiatr Genet 2012; 159B: 803–811.
Wade TD, Gordon S, Medland S, Bulik CM, Heath AC, Montgomery GW et al. Genetic variants associated with disordered eating. Int J Eat Disord 2013; 46: 594–608.
Boraska V, Franklin CS, Floyd JaB, Thornton LM, Huckins LM, Southam L et al. A genome-wide association study of anorexia nervosa. Mol Psychiatry 2014; 19: 1085–1094.
Guo Y, Wei Z, Keating BJ, Hakonarson H . Machine learning derived risk prediction of anorexia nervosa. BMC Med Genomics 2016; 9: 4.
Wang K, Zhang H, Bloss CS, Duvvuri V, Kaye W, Schork NJ et al. A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Mol Psychiatry 2011; 16: 949–959.
Nagashima T, Kim J, Li Q, Lydon JP, DeMayo FJ, Lyons KM et al. Connective tissue growth factor is required for normal follicle development and ovulation. Mol Endocrinol 2011; 25: 1740–1759.
Bulik CM, Reba L, Siega-Riz A-M, Reichborn-Kjennerud T . Anorexia nervosa: definition, epidemiology, and cycle of risk. Int J Eat Disord 2005; 37 (Suppl): S2–9–1.
Hosokawa M, Shoji M, Kitamura K, Tanaka T, Noce T, Chuma S et al. Tudor-related proteins TDRD1/MTR-1, TDRD6 and TDRD7/TRAP: domain composition, intracellular localization, and function in male germ cells in mice. Dev Biol 2007; 301: 38–52.
Katz MG, Vollenhoven B . The reproductive endocrine consequences of anorexia nervosa. BJOG 2000; 107: 707–713.
Smathers RL, Petersen DR . The human fatty acid-binding protein family: evolutionary divergences and functions. Hum Genomics 2011; 5: 170–191.
Crisp MS, Lane C, Halliwell M, Wynford-Thomas D, Ludgate M . Thyrotropin receptor transcripts in human adipose tissue. J Clin Endocrinol Metab 1997; 82: 2003–2005.
Haraguchi K, Shimura H, Lin L, Endo T, Onaya T . Differentiation of rat preadipocytes is accompanied by expression of thyrotropin receptors. Endocrinology 1996; 137: 3200–3205.
Lu M, Lin R-Y . TSH stimulates adipogenesis in mouse embryonic stem cells. J Endocrinol 2008; 196: 159–169.
Mullur R, Liu Y-Y, Brent GA . Thyroid hormone regulation of metabolism. Physiol Rev 2014; 94: 355–382.
Chen J, Ren J, Jing Q, Lu S, Zhang Y, Liu Y et al. TSH/TSHR Signaling Suppresses Fatty Acid Synthase (FASN) Expression in Adipocytes. J Cell Physiol 2015; 230: 2233–2239.
Lisowski P, Wieczorek M, Goscik J, Juszczak GR, Stankiewicz AM, Zwierzchowski L et al. Effects of chronic stress on prefrontal cortex transcriptome in mice displaying different genetic backgrounds. J Mol Neurosci 2013; 50: 33–57.
Cheung CYY, Tso AWK, Cheung BMY, Xu A, Ong KL, Fong CHY et al. Obesity susceptibility genetic variants identified from recent genome-wide association studies: implications in a chinese population. J Clin Endocrinol Metab 2010; 95: 1395–1403.
Bailer UF, Frank GK, Price JC, Meltzer CC, Becker C, Mathis CA et al. Interaction between serotonin transporter and dopamine D2/D3 receptor radioligand measures is associated with harm avoidant symptoms in anorexia and bulimia nervosa. Psychiatry Res 2013; 211: 160–168.
O’Hara CB, Keyes A, Renwick B, Leyton M, Campbell IC, Schmidt U . The effects of acute dopamine precursor depletion on the reinforcing value of exercise in anorexia nervosa. PLoS One 2016; 11: e0145894.
Frieling H, Römer KD, Scholz S, Mittelbach F, Wilhelm J, De Zwaan M et al. Epigenetic dysregulation of dopaminergic genes in eating disorders. Int J Eat Disord 2010; 43: 577–583.
Broft A, Slifstein M, Osborne J, Kothari P, Morim S, Shingleton R et al. Striatal dopamine type 2 receptor availability in anorexia nervosa. Psychiatry Res 2015; 233: 380–387.
Södersten P, Bergh C, Leon M, Zandian M . Dopamine and anorexia nervosa. Neurosci Biobehav Rev 2015; 60: 26–30.
Nagano M, Saitow F, Haneda E, Konishi S, Hayashi M, Suzuki H . Distribution and pharmacological characterization of primate NK-1 and NK-3 tachykinin receptors in the central nervous system of the rhesus monkey. Br J Pharmacol 2006; 147: 316–323.
Engman J, Åhs F, Furmark T, Linnman C, Pissiota A, Appel L et al. Age, sex and NK1 receptors in the human brain — a positron emission tomography study with [11C]GR205171. Eur Neuropsychopharmacol 2012; 22: 562–568.
Garcia-Recio S, Gascón P . Biological and pharmacological aspects of the NK1-receptor. Biomed Res Int 2015; 2015: 495704.
Muñoz M, Coveñas R . Involvement of substance P and the NK-1 receptor in human pathology. Amino Acids 2014; 46: 1727–1750.
Schank JR . The neurokinin-1 receptor in addictive processes. J Pharmacol Exp Ther 2014; 351: 2–8.
Sharp SI, McQuillin A, Marks M, Hunt SP, Stanford SC, Lydall GJ et al. Genetic association of the tachykinin receptor 1 TACR1 gene in bipolar disorder, attention deficit hyperactivity disorder, and the alcohol dependence syndrome. Am J Med Genet B Neuropsychiatr Genet 2014; 165B: 373–380.
Amoruso A, Bardelli C, Cattaneo CI, Fresu LG, Manzetti E, Brunelleschi S . Neurokinin (NK)-1 receptor expression in monocytes from bipolar disorder patients: a pilot study. J Affect Disord 2015; 178: 188–192.
Perlis RH, Purcell S, Fagerness J, Kirby A, Petryshen TL, Fan J et al. Family-based association study of lithium-related and other candidate genes in bipolar disorder. Arch Gen Psychiatry 2008; 65: 53–61.
Yan TC, McQuillin A, Thapar A, Asherson P, Hunt SP, Stanford SC et al. NK1 (TACR1) receptor gene ‘knockout’ mouse phenotype predicts genetic association with ADHD. J Psychopharmacol 2010; 24: 27–38.
Burnet PW, Harrison PJ . Substance P (NK1) receptors in the cingulate cortex in unipolar and bipolar mood disorder and schizophrenia. Biol Psychiatry 2000; 47: 80–83.
Frisch P, Bilkei-Gorzó A, Rácz I, Zimmer A . Modulation of the CRH system by substance P/NKA in an animal model of depression. Behav Brain Res 2010; 213: 103–108.
Brenes JC, Broiz AC, Bassi GS, Schwarting RKW, Brandão ML . Involvement of midbrain tectum neurokinin-mediated mechanisms in fear and anxiety. Braz J Med Biol Res 2012; 45: 349–356.
Carvalho MC, Santos JM, Bassi GS, Brandão ML . Participation of NK1 receptors of the amygdala on the processing of different types of fear. Neurobiol Learn Mem 2013; 102: 20–27.
Santarelli L, Gobbi G, Debs PC, Sibille ET, Blier P, Hen R et al. Genetic and pharmacological disruption of neurokinin 1 receptor function decreases anxiety-related behaviors and increases serotonergic function. Proc Natl Acad Sci U S A 2001; 98: 1912–1917.
Bassi GS, de Carvalho MC, Brandão ML . Effects of substance P and Sar-Met-SP, a NK1 agonist, in distinct amygdaloid nuclei on anxiety-like behavior in rats. Neurosci Lett 2014; 569: 121–125.
Ebner K, Singewald N . The role of substance P in stress and anxiety responses. Amino Acids 2006; 31: 251–272.
Yan TC, Hunt SP, Stanford SC . Behavioural and neurochemical abnormalities in mice lacking functional tachykinin-1 (NK1) receptors: A model of attention deficit hyperactivity disorder. Neuropharmacology 2009; 57: 627–635.
Porter AJ, Pillidge K, Tsai YC, Dudley JA, Hunt SP, Peirson SN et al. A lack of functional NK1 receptors explains most, but not all, abnormal behaviours of NK1R−/− mice 1. Genes, Brain Behav 2015; 14: 189–199.
Pillidge K, Heal DJ, Stanford SC . The NK1R−/− mouse phenotype suggests that small body size, with a sex- and diet-dependent excess in body mass and fat, are physical biomarkers for a human endophenotype with vulnerability to attention deficit hyperactivity disorder. J Psychopharmacol 2016; 30: 848–855.
Dando TM, Perry CM . Aprepitant: a review of its use in the prevention of chemotherapy-induced nausea and vomiting. Drugs 2004; 64: 777–794.
Ebner K, Sartori SB, Singewald N . Tachykinin receptors as therapeutic targets in stress-related disorders. Curr Pharm Des 2009; 15: 1647–1674.
Machado-Vieira R, Zarate CA . Proof of concept trials in bipolar disorder and major depressive disorder: a translational perspective in the search for improved treatments. Depress Anxiety 2011; 28: 267–281.
Olive MF . Neurokinin-1 (NK1 receptor antagonists as possible therapeutics for psychostimulant use disorders. CNS Neurol Disord Drug Targets 2015; 14: 700–706.
Murtra P, Sheasby AM, Hunt SP, De Felipe C . Rewarding effects of opiates are absent in mice lacking the receptor for substance P. Nature 2000; 405: 180–183.
Frank GKW . Altered brain reward circuits in eating disorders: chicken or egg? Curr Psychiatry Rep 2013; 15: 396.
Palamiuc L, Noble T, Witham E, Ratanpal H, Vaughan M, Srinivasan S . A tachykinin-like neuroendocrine signalling axis couples central serotonin action and nutrient sensing with peripheral lipid metabolism. Nat Commun 2017; 8: 14237.
Sergeyev V, Hökfelt T, Hurd Y . Serotonin and substance P co-exist in dorsal raphe neurons of the human brain. Neuroreport 1999; 10: 3967–3970.
Walker PD, Riley LA, Hart RP, Jonakait GM . Serotonin regulation of tachykinin biosynthesis in the rat neostriatum. Brain Res 1991; 546: 33–39.
Froger N, Gardier AM, Moratalla R, Alberti I, Lena I, Boni C et al. 5-hydroxytryptamine (5-HT)1A autoreceptor adaptive changes in substance P (neurokinin 1) receptor knock-out mice mimic antidepressant-induced desensitization. J Neurosci 2001; 21: 8188–8197.
Acknowledgements
We thank Dr Anna Di Nardo for gently collecting all skin biopsies used in this study. We thank Dr Brian Tobe for helpful discussions and critiques on using stem cells in psychiatry. The Muotri lab is supported by grants from the California Institute for Regenerative Medicine (CIRM) TR4-06747, the National Institutes of Health through the NIH Director's New Innovator Award Program (1-DP2-OD006495-01), an R21 1R21MH093954; and a NARSAD Independent Investigator Grant to ARM.
Author contributions
ARM and PDN designed the experiments with input from all authors and wrote the manuscript. PDN, FRC and TC generated and characterized the iPSCs. PDN and FRC generated and characterized NPCs and neuronal populations. PDN performed all gene expression/protein assays and statistical analysis with help from FRC. CAT performed PCR array analysis. RHH and ASC performed all bioinformatics analysis. VD helped to design the experiments and analysis, and co-wrote the manuscript with ARM and PDN. All authors reviewed the manuscript for publication.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Negraes, P., Cugola, F., Herai, R. et al. Modeling anorexia nervosa: transcriptional insights from human iPSC-derived neurons. Transl Psychiatry 7, e1060 (2017). https://doi.org/10.1038/tp.2017.37
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2017.37
This article is cited by
-
Brain-based gene expression of putative risk genes for anorexia nervosa
Molecular Psychiatry (2023)
-
Molecular neuroanatomy of anorexia nervosa
Scientific Reports (2020)
-
Mental health dished up—the use of iPSC models in neuropsychiatric research
Journal of Neural Transmission (2020)
-
Tissue-type plasminogen activator-primed human iPSC-derived neural progenitor cells promote motor recovery after severe spinal cord injury
Scientific Reports (2019)
-
Neural stem cells and epilepsy: functional roles and disease-in-a-dish models
Cell and Tissue Research (2018)
