
Abstract
Tubby and Tubby-like proteins (TLPs), in mammals, play critical roles in neural development, while its function in plants is largely unknown. We previously demonstrated that the chickpea TLP, CaTLP1, participates in osmotic stress response and might be associated with ABA-dependent network. However, how CaTLP1 is connected to ABA signaling remains unclear. The CaTLP1 was found to be engaged in ABA-mediated gene expression and stomatal closure. Complementation of the yeast yap1 mutant with CaTLP1 revealed its role in ROS scavenging. Furthermore, complementation of Arabidopsis attlp2 mutant displayed enhanced stress tolerance, indicating the functional conservation of TLPs across the species. The presence of ABA-responsive element along with other motifs in the proximal promoter regions of TLPs firmly established their involvement in stress signalling pathways. The CaTLP1 promoter driven GUS expression was restricted to the vegetative organs, especially stem and rosette leaves. Global protein expression profiling of wild-type, attlp2 and complemented Arabidopsis plants revealed 95 differentially expressed proteins, presumably involved in maintaining physiological and biological processes under dehydration. Immunoprecipitation assay revealed that protein kinases are most likely to interact with CaTLP1. This study provides the first demonstration that the TLPs act as module for ABA-mediated stomatal closure possibly via interaction with protein kinase.
Similar content being viewed by others

Introduction
Plants respond and adapt to water-deficit or dehydration conditions by altering their architecture, cellular metabolism and activating various defense machineries. Such responses to dehydration are regulated by differential expression of many genes/gene-products, which are involved in a number of different but potentially overlapping, signal transduction pathways. Analysing the functions of stress-responsive genes is critical to better understand the molecular mechanism governing stress response, which ultimately opens up new avenues for improving stress tolerance. The past decades have seen the prospect of genetically modified plants and testing their potential to modulate tolerance through alteration of the osmolyte levels and enzymes that scavenge reactive oxygen species (ROS), components involved in signal transduction, transcription factors (TFs) and regulators.
Abscisic acid (ABA) level, under stress conditions, increases dynamically in vegetative organs, triggering adaptive responses that are essential for plant survival and productivity1. It induces stomatal closure and minimizes water loss through transpiration. On the contrary, high levels of ABA cease overall plant growth2. Numerous genes have been reported that display deregulated expression under stress conditions, and many of them are mediated by ABA3,4. This expression is modulated by the binding of various TFs onto cis-regulatory regions of a gene, which act as molecular switches controlling various biological processes including abiotic stress and hormone responses. The TFs have proven quite useful in improving stress tolerance in transgenic plants, through influencing expression of a number of stress-responsive genes5,6.
The tubby proteins are encoded by a multigene family. The first tubby gene was identified in mice, and shown to be associated with late-onset obesity and neurosensory deficits7. The proteins that are homologous to tubby are known as tubby-like proteins (TLPs). The members of tubby family have been found to perform multiple roles including vesicular trafficking, insulin signalling, gene transcription, G-protein signalling, and ribosomal RNA synthesis8, among others. Tubby proteins are present in organisms ranging from unicellular to multicellular eukaryotes9. While in mammals, these proteins form a small family, which consists of Tubby, and four TLPs10, plants appear to harbor a large number of TLPs9,10,11. This expansion is the result of segmental duplication and random translocation and insertion12 indicating a significant role of TLPs in plants. However, exploration of the molecular function of these proteins remains still elusive. Recent studies have explored the role of TLPs in abiotic stress signalling9, and patho-stress11,13. In a previous proteomics study, we identified the chickpea TLP, designated CaTLP1 in the extracellular matrix14, and demonstrated that CaTLP1 is a putative transcription factor presumably involved in multivariate stress15.
Biological processes are executed by proteins that, to a large extent, depend on interactions with other proteins for their activity. These interactions are specific, even among members of a particular protein family that contain similar interaction domains. Studying these specific interactions reveals the molecular network that may lead to potential functional linkages and molecular explanations of biological processes in an organism. Several previous studies established the significant findings in the study of protein complexes formed by ectopically overexpressed proteins from same as well as other plant species16,17. To date, generation of large-scale protein-protein interaction (PPI) maps has relied on the yeast two-hybrid system, which detects binary interactions through activation of a reporter gene expression18. However, ultrasensitive mass spectrometric identification is capable of directly identifying the protein complexes on a proteome-wide scale19. The advantage of analyzing purified protein complexes is the ability to identify specific interacting proteins and post-translational modifications that may otherwise go undetected in large-scale global analyses. This is achieved by performing an immunoprecipitation with a bait protein and analyzing the prey proteins using mass spectrometry20. Insight into individual pathways can be obtained by aiming a particular group of proteins that is reported to be enriched for interactions. Interactions of TFs increase the selectivity of protein-DNA interactions and create a large number of diverse DNA-binding complexes from a relatively small number of proteins. In this study, we investigated the potential role of CaTLP1 in modulating plant responses to abiotic stress using mutant, complemented and overexpressing lines of Arabidopsis. We showed that the CaTLP1 equips plants to withstand such stress through stomatal modulations and regulation of gene expression via ABA-dependent pathways. Further, we used mass spectrometry based interactive proteomics for the identification of CaTLP1-complex components that mediate stress tolerance.
Results
CaTLP1 counteracts oxidative stress in yeast
In a previous study, we demonstrated that CaTLP1 could impart improved tolerance against multivariate stresses15, possibly through regulation of antioxidant defense genes. Detoxification of ROS is a common mechanism of plant adaptation or tolerance to oxidative stress. The budding yeast, S. cerevisiae harbors the same oxidative stress defense mechanisms as in higher eukaryotes21 and therefore, to examine the precise role of CaTLP1 in ROS-scavenging, functional complementation was carried out in yeast mutants viz. yap1, sod1 and trx1/trx2 affected in antioxidant function. The coding sequence of CaTLP1 was cloned in expression vector pYES2 and introduced in yap1, sod1 and trx1/trx2 in the BY4741 background. The transformants were selected on auxotrophic media and tested for their growth phenotype on YPD media containing various stress-inducing chemicals (0.5 mM menadione, 1.2 M NaCl, 5 mM H2O2 and 0.1 M LiCl). While there was inhibition of growth under menadione and H2O2 treatments, the growth phenotype of the mutant in the plate containing other stressors was not very significant from that of WT strains (Supplemental Fig. 1a). However, the OD based observation in liquid media indicated that expression of CaTLP1 could enhance the survival of yap1 when compared with sod1 and trx1/trx2 mutants (Fig. 1). The mutant strains transformed with empty vector could not rescue the growth defect. These results revealed that CaTLP1 could rescue the suppressed activity in the yeast mutants, suggesting its key role in regulation of ROS.

Heterologous complementation of yeast mutants by CaTLP1.
Wild-type and yap1 (a,d,g,j) sod1 (b,e,h,k), trx1/trx2 (c,f,i,l) yeast mutants, transformed with pYES2 and pYES2-CaTLP1, were cultured in SD-URA media overnight and diluted in fresh media (approximate OD600 value of 0.01) before being subjected to (a–c) 5 mM H2O2, (d–f) 1.2 M NaCl, (g–i) 0.1 M LiCl and (j-l) 0.5 mM menadione treatments. The cultures were incubated at 30 °C and OD600 readings were taken at every 1 h. Data shown are the means ± SD of three biological replicates.
Expression of CaTLP1 alters stress response in transgenic Arabidopsis
To explore in planta function of CaTLP1 in stress response, we complemented its homolog in Arabidopsis. When searched against TAIR database, AtTLP2 was found to be the closest ortholog of CaTLP1 in Arabidopsis. T-DNA mutant (SALK_058100 C) was selected as null mutant and complemented lines with CaTLP1 were generated. On an average, 4–6 homozygous lines, both complemented and overexpressing transgenics, were obtained in T4 generation. The wild-type, CaTLP1 overexpressing line (OE-11) and complemented plants (OEC-3, OEC-6) along with attlp2 mutant were selected for further studies.
The transgenic plants were tested for their seed germination and survival under stress conditions. Consistent with our previous results15, the overexpressing plants showed better phenotype as compared to mutants and wild-type counterparts (Fig. 2). The mutants did not show any difference in germination rate as compared to wild-type in unstressed control condition. However, under hypersalinity, osmotic and oxidative stress conditions, the germination rate of CaTLP1 complemented plants were much higher than mutants and wild-type (Fig. 2a,b), indicating early response of CaTLP1 in stress signaling. In a separate set of experiment, the four-leaf stage seedlings were transferred to pots and allowed to grow further. The 3-week-old transgenic plants harboring CaTLP1 showed better growth as compared to the mutants (Fig. 2c). When subjected to stress conditions, the mature transgenic plants exhibited enhanced tolerance to dehydration (Fig. 2d). In parallel, a separate set of pots were kept well-watered. Apart from the complemented lines, there were no significant differences in the phenotype of plants under unstressed condition (Supplemental Fig. 1b). However, the reduction in biomass of complemented line under dehydration was significantly lower as compared to wild type and mutant (Supplemental Fig 2 and Supplemental Table 1). The alteration in root length was monitored on MS plates supplemented with various stressors viz. NaCl for hypersalinity, H2O2 for oxidative stress and mannitol for dehydration, besides ABA treatment. The transgenic seedlings showed less inhibitory effects on root growth as compared to wild-type and attlp2 mutants (Fig. 2e,f and Table 1). When subjected to ABA treatment, the degree of inhibition in attlp2 mutants and wild-type was 47 and 33%, respectively as against 28% in overexpressing and 17–23% in complemented lines. The reduction in root length under hypersalinity was 59 and 34% in mutant and wild-type, respectively whereas it was 16–28% in transgenics indicating their better adaptability to such conditions (Fig. 2g).

Effect of stress on different stages of wild-type, mutant and transgenic Arabidopsis.
(a) Germination assay of wild-type, attlp2, complemented (OEC-3, OEC-6) and overexpressing (OE-11) line(s) on MS media supplemented with H2O2 (4 mM), ABA (4 μM), and NaCl (200 mM). (b) Seed germination percentage under various stresses. (c) Morphology of 3-week-old seedling of the indicated lines. (d) Morphological features of 4-week-old plants of the indicated lines subjected to dehydration for 10 d. (e) Seedlings at four-leaf stage were transferred to various stresses as indicated and growth was monitored after 7 d. (f) Histogram showing the differential root length. (g) Percent reduction in root growth of the indicated lines.
CaTLP1 expression influences stomatal movements
Stomatal closure is the most rapid response at the onset of water-deficit condition followed by, on a longer time scale, reduced plant growth and decreasing transpiration thereby facilitating conservation of water22. Therefore, we examined the effect of CaTLP1 on stomatal movements. Under unstressed control condition, the stomata in wild-type, mutant and transgenic plants remained open (Fig. 3a). The size of leaf epidermal cells in the transgenics was larger than the mutants and wild-type plants. These results are in consistent with our previous observation where CaTLP1 overexpressing tobacco plants displayed better stress adaptation by maintaining higher photosynthetic rate and reduced water loss15. Upon treatment with dehydration, the overexpressing and complemented plants showed inhibition of stomatal opening by 75–80% as against 40–50% in wild-type and 20–30% in mutants (Fig. 3b). Examination of stomatal aperture in leaves under dehydration showed that, relative to wild-type plants, CaTLP1 overexpressing plants had smaller stomatal apertures and attlp2 mutants had larger stomatal apertures (Fig. 3c,d). These results suggest that the overexpression of CaTLP1 might be involved in stomatal movement, which affect guard cells morphology, and in turn lead to better adaptation.

Effect of CaTLP1 on stomatal movement.
(a) Four-week-old attlp2, wild-type and transgenic plants (OEC-3, OEC-6 and OE-11) were uprooted and subjected to dehydration stress. Scanning electron micrographs display the changes of stomatal movement in transgenics with respect to mutant and wild-type plants. The bar indicates the extent of resolution. (b) Percentage reduction in number of opened stomata (aperture ≥2 μm). (c) Reduction in the stomatal aperture in response to dehydration stress. (d) Histogram shows the percent reduction in stomatal aperture for the indicated lines. The results are the means ± SE for 120 stomata. Transcript abundance of stress-responsive genes viz., RD20 (e), RAB18 (f), bZIP20 (g), polyubiquitin (h) and GroEL (i) were quantified. Quantitative real time PCR was performed using gene-specific primers (Supplemental Table 5). The relative gene expression was evaluated using comparative cycle threshold method with EF-1α as the reference gene. Data shown are the means ± SD of three biological replicates.
Overexpression of CaTLP1 induces stress-responsive genes
The positive effect of CaTLP1 overexpression on abiotic stress tolerance prompted us to check the transcript abundance of stress-responsive marker genes. When subjected to multivariate stress conditions, the expression of RD20, RAB18, GroEL, polyubiquitin and bZIP20 in the transgenic plants was significantly different from that in the mutants and wild-type (Fig. 3e–i). The comparison showed early inductions of RD20, RAB18, and GroEL, while polyubiquitin expression was delayed in the transgenics. The expression pattern of GroEL was found to be opposite to that of polyubiquitin. Under the same experimental conditions, the transcripts of bZIP20 were highly induced in the mutants when compared with wild-type and transgenics. These results altogether suggest a possible role of CaTLP1 in the regulation of expression of downstream stress-responsive genes.
Expression of CaTLP1 influences proteome profiles
Next we investigated whether expression of CaTLP1 has any influence on the proteome profiles of wild-type, mutants and complemented plants. Three replicate 2-DE gels of each line were computationally combined into representative standard gels. The proteome of wild-type, mutant and the representative complemented line (OEC-6) displayed 195, 164 and 240 protein spots, respectively. A second level matchset was then created (Fig. 4a). The intensity of spots was normalized to that of landmark protein spots used for internal standardization. The filtered spot quantities were assembled into a data matrix, which consisted of 390 spots, indicating change in intensity for each spot. Approximately 94% of the spots on the standard gels were of high quality, reflecting the reproducibility of the replicates. Quantitative image analysis revealed a total of 95 protein spots that changed their intensities significantly by more than 2.5-fold. The differentially regulated proteins in attlp2 and OEC-6 are represented by Venn diagram (Fig. 4b).

Comparative proteome profiling displaying differentially expressed proteins.
(a) The higher level matchset was created in silico from three standard gels for each of the indicated lines. The numbers were assigned to the spots by PDQuest software (version 7. 2. 0). (b) Venn diagram showing the distribution of differentially expressed proteins in attlp2 and complemented (OEC-6) lines as against wild-type plants.
CaTLP1 promoter harbors stress-responsive regulatory elements
Since CaTLP1 was found to have a regulatory role in stress tolerance, we performed an in silico analysis of CaTLP promoter region. A 1.4 kb 5′-upstream region of CaTLP1 was amplified by genome walking approach. The 5′-upstream regions of two paralogs of CaTLP, designated CaTLP2 and CaTLP3, were retrieved from chickpea draft sequence23. Using PLACE and PLANTCARE databases, we analysed the sequences and predicted the key cis-acting elements such as ABRE, MYBPLANT, MYCCONSENSUS and ROOTMOTIF, among others (Supplemental Table 2), albeit the number of predicted cis-elements differed in the paralogs. The analysis revealed that most elements in CaTLP promoters were mainly environment- or hormone-responsive. A putative TATA box motif was found at 198 bp upstream of the CaTLP1 ORF, while O2-site and G-box sequence was found at 228 bp and 408 bp, respectively. G-box element was found with Box-1 and Box-4 at the upstream of the CaTLP2 and CaTLP3 ORFs, respectively. These motifs along with the ABRE form various types of ABRE cassettes, which help in effective regulation by ABRE motif itself. Interestingly, ABRE motif was present in CaTLP1 and CaTLP2, but not in CaTLP3 (Fig. 5a), suggesting the different functions of TLPs in chickpea. We, therefore, propose that CaTLP1 promoter is an inducible promoter and might be regulated by multiple abiotic stress factors and hormones.

Proximal regulatory elements of CaTLP1 and influence on ABA-mediated gene expression.
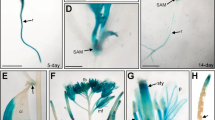
(a) Comparison of regulatory elements in the proximal promoter region of chickpea TLPs. (b) Expression of ProCaTLP1-GUS in Arabidopsis. GUS expression is shown in root (I), epicotyl (IV) and leaf (VII) of wild-type; root (II), epicotyl (V) and leaf (VIII) of 10-d-old transgenic seedlings; root (III), stem (VI) and leaf (IX) of 6-week-old transgenic plants. Transcript accumulation of GUS under progressive dehydration (c) and various other stresses (d). Data shown are the means ± SD of three biological replicates.
The genomic sequence upstream of the CaTLP1 orthologues in other plant species were retrieved from the NCBI and Phytozome database (http://www.phytozome.net/) and subjected to comparative analysis by multiple alignment with ClustalW and PlantCARE programs. The analyses revealed the presence of consensus ABRE sequences (Supplemental Table 3) with several common cis-acting elements in most orthologs.
CaTLP1 is predominantly expressed in vegetative organs
To study the tissue-specific expression and transcriptional regulation of CaTLP1, we developed ProCaTLP1:GUS lines in Arabidopsis. Tissue samples from 6-d-old seedlings and 4-week-old plants were analyzed for GUS-staining. The fusion protein showed distinct expression profile across developing stages (Fig. 5b). GUS-staining was detected in roots, leaves and epicotyls of young seedlings, and was significantly higher in mature vegetative organs, especially stem and rosette leaves but not in roots. Interestingly, promoter activity was observed predominantly in leaf blade of matured plants, whereas in seedlings, maximal activity was restricted to the leaf edges. These results indicate overlapping but distinct expression pattern of CaTLP1 during developmental stages.
ABRE regulon of CaTLP1 is involved in osmotic stress responses
Since ABRE is known as a cis-element involved in ABA signaling, we surmised that the ABRE sequence in the CaTLP1 promoter might be involved in ABA-mediated signaling. The 10-d-old seedling expressing ProCaTLP1-GUS were grown on MS media supplemented with ABA and other stressors viz. H2O2, mannitol and NaCl in various concentrations for 5 days, and the transcript abundance of GUS was quantified. The transcript level increased 2- to 9-fold upon dehydration for 5 h (Fig. 5c), and 3-, 8- and 6-fold with H2O2, NaCl and mannitol treatments, respectively (Fig. 5d). The accumulation of transcripts was much higher upon treatment with 2 and 4 μM ABA (approximately 9- and 25-fold, respectively). These results indicate that the ABRE sequence might be responsible for the osmotic stress induced expression of CaTLP1 via ABA-dependent pathway.
CaTLP1 complexes with kinase family proteins as interacting partners
Our findings demonstrated that CaTLP1 plays a key role in multivariate stress responses. To address how CaTLP1 responds to stress adaptation, we investigated the interactome using transgenics overexpressing CaTLP1-FLAG fusion protein (OE-11). The protein complex from 5-week-old transgenic plants was immunoprecipitated using anti-FLAG antibody. Similarly anti-CaTLP1 antibody was used to immunoprecipitate CaTLP1-protein complex from 4-week-old chickpea plants. The immunoprecipitates were separated by SDS-PAGE, followed by detection with CBB staining (Fig. 6a). The purity of immunoprecipitates was confirmed by anti-Rubisco antibody (Fig. 6b). Protein band corresponding to 47 kDa, from purified fraction, indicated the presence of CaTLP1 which was further confirmed by immunoblot analysis. The immunoprecipitates displayed few extra bands in both chickpea and transgenic Arabidopsis, indicating possible interacting proteins. Additionally, immunoblot analyses with anti-FLAG and anti-CaTLP1 antibodies confirmed the presence of CaTLP1 in the immunoprecipitates (Fig. 6c and d).

Putative interacting partners of CaTLPs.
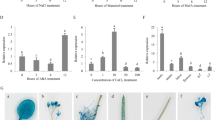
(a) CBB-stained SDS-PAGE gel showing protein bands immunoprecipitated from transgenic Arabidopsis (OE-11) and chickpea, probed with anti-FLAG and anti-CaTLP1 antibodies, respectively. (b) Western blot analysis of immunoprecipitated proteins with anti-Rubisco antibody. (c,d) Confirmation of immunoprecipitates using anti-FLAG and anti-CaTLP1 antibodies. CC, crude protein from chickpea; CA, crude protein from Arabidopsis; P, purified protein; PA, purified protein from Arabidopsis; PC, purified protein from chickpea; W, wash fraction; PWT, purified protein from wild-type plants. Arrows indicate band of purified CaTLP1. (e) Surface view interaction of F-Box domain (purple) of CaTLP1 and CaTLP2 (green) with casein kinase 1-like protein 2 (AtCK2) and CaCK1 (cyan) visualized with PyMOL. (f) Interactions of CaTLP1 and CaTLP2 with kinase family proteins showing common as well as exclusive members in the association network. (g) AtTLP2 interaction showing the common casein kinase family protein CK (pink circle) in the network. (h) BiFC assay showing interaction of nYFP-CaTLP1 and cYFP-CaCK1.
Next we performed mass spectrometry analysis to identify the proteins present in the immunoprecipitates using LC-ESI/MS/MS. A total of 12 proteins were identified in the immunoprecipitates. A critical screening revealed 10 proteins to be potential interacting partners, most of which were from the protein kinase family (Supplemental Table 4). Casein kinase 1-like protein 2 (CK2) from Arabidopsis and protein kinase from chickpea exhibited the highest Mascot score, indicating most likeliness to interact with CaTLP1.
CaTLP1 and CaTLP2 interact with protein kinases through F-box
Previous studies demonstrated the interaction of casein kinase with proteins harboring F-box domain24,25. Thus, we performed computational docking experiments to predict the contact interface of CaTLP1 and CaTLP2 with Arabidopsis CK2 and chickpea protein kinase (designated CaCK1). The results suggest that CK2 and CaCK1 interact with CaTLPs through F-box domain (Fig. 6e). To further validate these interactions, system biology approaches were applied to construct the phosphorylation oriented protein kinase interactome. The individual interactions of CaTLP1 or CaTLP2 along with common interactions of the CaTLPs are shown in Fig. 6f. Among 55 interactions, 5 were unique to CaTLP1, 22 were unique to CaTLP2, while 26 interactions were shared by both the TLPs. The two categories of interacting partners shown by the protein kinase interactome suggest the interaction diversity between the two paralogs. AtTLP2 was also predicted to interact with casein kinase and protein kinase family (Fig. 6g). To further validate the interaction between CaTLP1 and CaCK1 in planta, a bimolecular fluorescence complementation (BiFC) was performed in tobacco leaf epidermal cells. In this assay, putative partners are fused to inactive N-terminal and C-terminal moieties of YFP. Bait and prey association brings YFP fragments together and drives reassembly of the active fluorescent protein, providing a direct readout of the interaction26. In the experiment with CaTLP1 + CaCK1, one out of eight combinations gave positive signal i.e. YFP fluorescence, in transfected tissues (Fig. 6h). These results reconfirm that CaTLP1 physically interacts with CaCK1 in vivo. Although, CaTLP2 was predicted to interact with protein kinase family and other various classes of kinases, no interaction could be predicted with casein kinases. These results indicate specific interactions and thus distinct functions of various TLPs.
Discussion
Water deficiency in soil interferes with mineral nutrition and water uptake, which leads to accumulation of toxic ions in plants. To reduce these detrimental effects, plants use several strategies, including the regulated expression of specific proteins, which eventually re-establishes proper cellular ion and osmotic homeostasis with other concomitant processes of repair and detoxification27. The machinery leading to the expression of dehydration-responsive genes conforms to the general cellular model, with a complex signal transduction cascade that can be divided into the following steps: (a) perception of stimulus; (b) processing, including amplification and integration of the signal; and (c) a response reaction in the form of de novo gene expression.
Kamada et al.28 proposed an attractive model for the stress-induced activation of a transduction pathway by investigating the heat-shock response in yeast, which may provide the conceptual framework for devising experiments in plants. The exposure of yeast cells to low doses of H2O2 or O2-generating drugs switches on within minutes a resistance to toxic doses of these oxidants29. The redox-oxidized ROS balance has been compared in a range of yeast mutants. These include the genes affecting antioxidant function at three distinct levels: transcription factors that induce expression of oxidative stress-responsive genes (Yap1), defence enzyme encoding superoxide dismutase (Sod1), and protein disulfide bond metabolism (Trx1 and Trx2). Mutants deficient in Yap1p show reduced activities of several enzymes with antioxidant activity such as SOD, glucose-6-phosphate dehydrogenase, and glutathione reductase30. The adaptive response to H2O2 is also severely affected by yap1 mutants, suggesting that Yap1p affects the transcription of genes involved in such response31. It is of significance that the transcription of TRX2 is severely impaired by Yap1 mutation, yet the TRX2 transcription is induced by H2O2, indicating that additional factor(s) might be involved in this process32. Yap1 controls a large oxidative stress-responsive regulon, which includes most of the oxidant scavenging enzymes Sod1p and Sod2p. Its function can be activated by several chemical stressors including H2O2 and menadione33, and this activation is attributed to a redox stress-imposed nuclear redistribution of the protein. In view of the above, yap1 mutant was functionally complemented with CaTLP1, which could enhance its survival (Fig. 1) suggesting a putative role of CaTLP1 in ROS regulation.
Among other responses common in many distinct abiotic stresses, is the induction of cellular architectural changes. Mechanistically distinct abiotic stresses can all give rise to alterations in plant morphology. In this study, we observed that the root growth of CaTLP1 transgenics was much better than mutant and wild-type, when subjected to various stresses (Fig. 2). It is generally believed and supported by correlative evidence that a larger root system might be advantageous to cope with water-deficit conditions. This view has been underpinned by a comparison of near-isogenic lines that differed essentially in the size of their root system34. In many circumstances, it is the stress sensitivity of the root that limits the productivity35. An improved understanding of molecular responses of roots to such abiotic stresses may therefore facilitate the development of crops with increased tolerance.
During dehydration, stomata are induced to close as leaves sense water-deficit, particularly after the leaf water potential drops below certain threshold level. The production of ABA triggers the increase of cytosolic Ca2+ concentration in guard cells both via IP3 signal transduction cascade and also, via cADPR. These processes are correlated with a reduction in stomatal aperture36. It is well established that ABA promotes water-deficit tolerance in plants through stomatal regulation37. In this context, CaTLP1 might function as a positive regulator of ABA-mediated stomatal closure. To activate stress responses, genetic reprogramming is required, and several hundred genes have thus far been identified as stress-responsive38,39. Among the early induced genes, RD20, a member of caleosin appears to be one of the most highly expressed and is often used as a stress marker40. CaTLP1 could modulate the expression of several of the marker genes, suggesting that it might play a regulatory role for downstream stress signaling possibly by controlling guard cell morphology (Fig. 3).
Comparative proteomics has previously been used to decipher the differential protein expression in genetically modified plants41,42. In our investigation, transgenic Arabidopsis (OEC-6) and their isogenic controls (attlp2 mutant and wild-type) were grown side-by-side in environmentally controlled conditions. By cross-comparing the proteome profiles of these lines, we were able to eliminate the natural variability as well as the environmental effects and determine the possible effects related to differential gene expression as a consequence of transgene expression. Previous studies suggested that the differences in spot quantity between transgenic and non-transgenic lines fell in the range of natural variation or were part of the intended effects43,44. However, the fact that quite a high number of protein spots were found to be significantly modulated in CaTLP1 transgenics (Fig. 4) is in consistence with the results found in the previous proteomics studies of transgenic maize41 and potato45.
The promoter sequence of TLPs harbors multiple stress-responsive cis-acting elements (Fig. 5). It is interesting that the cis-acting elements were nearly identical, despite their low homology. The comparison of the CaTLP1 promoter with its orthologs in other eudicot species revealed a conserved ABRE motif, which might have a crucial role in its function. The regulation of ABA- and/or stress-responsive gene expression by ABRE and their putative cognate trans-acting factors have been studied extensively46,47. The CaTLP1 promoter contains other cis-elements such as O2-site and G-box besides ABRE (Supplemental Table 2), which are known to be involved in ABA-induced gene expression, and implicated in stress response. Differential expression profiles of GUS under CaTLP1 promoter indicated that there exists differential transcriptional regulation of CaTLP1 across the developmental stages and in response to environmental conditions (Fig. 5). Furthermore, several homologous regulatory motifs were identified which could be recognised by MYB, MYC or ABRE binding factors. It is thus conjectured that CaTLP1 has specific regulatory system similar to the genes having ABA inducible promoters, which might play a key role in the regulation of expression of CaTLP1 promoter.
Protein interactions that are clustered based on similar interaction patterns can serve as backbones for more complex molecular networks responsible for a particular function or pathway16,17. We identified a number of CaTLP1 interacting partners that include Arabidopsis CK2 and chickpea protein kinase CaCK1 (Supplemental Table 4). Numerous kinases and phosphatases are known to form distinguishable complexes often with regulatory factors that serve to localize or control activity48. The available data indicate that ABA or abiotic stresses induce several TFs, for example, ABF/AREB by triggering their phosphorylation49. This phosphorylation of TFs is necessary to induce downstream genes and could occur on the casein kinase II (CKII) phosphorylation sites present in the conserved domains50. Lai et al.9 demonstrated the physical interaction between AtTLP9 and ASK1 (Arabidopsis Skp1 like 1) through F-box, which may participate in the ABA signalling pathway during seed germination and early development. It is likely that CaTLP1 harboring F-box might play key role in stress tolerance through reprogramming of downstream stress-responsive genes.
We propose that CaTLP1 may be associated with synaptic function in stress tolerance (Fig. 7). The presence of ABRE in CaTLP1 promoter suggests the regulation of its expression in an ABA-dependent manner. CaTLP1 comprises two characteristics domains, Tub and F-box and is localized in the extracellular matrix, presumably serving as a molecular sensor for various stress signals. Upon perceiving such signals, it translocates to the nucleus. The translocation might be facilitated by its interaction with CaCK1 protein since casein kinase 2 family proteins are known to translocate extracellular signal-regulated proteins to the nucleus51. After being translocated to the nucleus, CaTLP1 might bind to DNA and interact with the regulator bound upstream to the target genes with the help of Tub and F-box domains, respectively. F-box domain either may regulate the activity or recruit other regulators at the promoter that can effectively switch on and off downstream gene expression. The mode of action of these genes is currently unknown, but they might contribute to decreased transpiration through effective stomatal closure and increased rate of photosynthesis under stress conditions. However, under unstressed conditions, these genes render support in cellular development by means of their effect on root and shoot development. Our future efforts would focus on exploring precise mode of functioning and downstream target genes of CaTLP1 in stress response, and to evaluate its use as modifier of plant development and stress tolerance.

Hypothetical model depicting the possible mechanism of action of CaTLP1.
Stress mediated ABA-dependent expression of CaTLP1 (1-4); feed-back inhibition of CaTLP1 (5), perception of signal by CaTLP1 (6); interaction of CaTLP1 with CaCK1 in either possible way (7), nuclear translocation (8); interaction with the upstream regulator (9); and switching on and off the downstream genes (10A and 10B) as detailed in the text. TUB, Tub domain; F-box, F-box domain; R, regulator.
Methods
Yeast complementation assay
The yeast expression vectors were constructed and transformation was carried out as described previously52. The CaTLP1 coding region was cloned in pYES2 vector (Invitrogen) using BamH1/Not1 restriction sites. The wild-type BY4741 (MATa; his3Δ 1; leu2Δ 0; met15Δ 0; ura3Δ 0) and the mutants yap1 (MATa; his3Δ 1; leu2Δ 0; met15Δ 0; ura3Δ 0 YML007w::kanMax4), sod1 (MATa; his3Δ 1; leu2Δ 0; met15Δ 0; ura3Δ 0 YJR104c::kanMax4) and trx1/trx2 (MATa; his3Δ 1; leu2Δ 0; met15Δ 0; ura3Δ 0 YGR209c::kanMax4) were transformed with pYES2:CaTLP1 or the empty vector pYES2. Growth assays were performed by inoculating wild-type and transformed strains in the respective medium supplemented with 0.5 mM menadione, 1.2 M NaCl, 5 mM H2O2 and 0.1 M LiCl. The overnight cultures were diluted to OD600 of 0.01 in the respective medium, and growth was monitored at every 1 h at 30 °C.
Plasmid construction and transformation
The overexpressing CaTLP1 binary vectors were constructed using the full-length cDNA of CaTLP1. The cDNA as well as 1.4 kb 5′ upstream sequence were cloned independently into pENTR/D-TOPO vector (Invitrogen). The cDNA was subcloned into the Gateway destination vectors, pGWB411. The CaTLP1 promoter was cloned into destination vector pGWB433 containing C-terminal GUS reporter gene. The gateway cloning was performed using LR Clonase (Invitrogen). The expression plasmids were transformed into Agrobacterium strain GV3101.
Plant materials and growth conditions
Seeds of wild-type (Col-0) and T-DNA mutants were obtained from The Arabidopsis Information Resource (TAIR), Carnegie Institution for Science, Department of Plant Biology, Stanford, USA. The seedlings were grown on Pro-Mix soil (Premier Horticulture) in a growth chamber under ambient humidity53. Plants were illuminated by Vita-lite fluorescent lamps (Durotest, Fairfield NJ) between 150 and 200 μmol m−2 s−1 of PAR at 16 h photoperiod. The plants were transformed by the floral-dip method, as previously described54 and homozygous seeds were generated for transgenic lines.
Stress and ABA treatment
Germination assay was performed using approximately 200 surface-sterilized seeds for each line, plated on MS medium without or supplemented with H2O2 (4 mM), NaCl (150 mM), and ABA (4 μM) and kept at 21 °C with 16 h photoperiod, and rate of germinated seeds were scored daily. Four-leaf staged seedlings (principal growth stage: 1.02–1.03) were grown on MS plates supplemented with H2O2 (2 and 4 mM), mannitol (200 mM), NaCl (200 mM), and ABA (2 and 4 μM). The seedlings were incubated for 7 d and phenotypic changes were recorded. Primary root length was measured on scanned images using ImageJ software (http://rsbweb.nih.gov/ij). In a separate experiment, 3-week-old plants grown in pots were subjected to dehydration by withdrawing water. Samples were collected and stored at −80 °C unless described otherwise.
Nucleic acid isolation and analysis
Genomic DNA was isolated using phenol extraction method (http://www.biotech.wisc.edu/ NewServicesandResearch/Arabidopsis/). The fragment flanking the T-DNA insert was amplified by PCR using appropriate primers (Supplemental Table 5). RNA was extracted using the TriPURE reagent (Molecular Research Center) and reverse transcribed into cDNA using a poly (dT) reverse primer and Superscript II reverse transcriptase (Invitrogen) after DNase treatment (Roche Diagnostics). The qRT-PCR assays were performed with the ABI PRISM 7700 sequence detection system (Applied Biosystems) using SYBR Green PCR Master mix including cDNA template and appropriate primers (Supplemental Table 5). The internal standard EF-1α was used for normalizing the qRT-PCR data and quantification was conducted as described previously55.
Protein extraction and 2-DE
Total proteins were extracted from 4-week-old plants as described earlier56. The protein extracts were dissolved in 200 μL rehydration buffer containing 2% immobilized pH gradient (IPG) buffer (pH 4–7), and quantified using 2-D Quant kit (GE Healthcare).
Isoelectric focusing (IEF) was performed with 250 μg proteins per IPG strip (13 cm, pH 4–7) using IPGphor system (GE Healthcare) as described57. The strips were then placed on top of 12.5% SDS-PAGE for second-dimension separation at a constant voltage of 100 V. The electrophoresed proteins were visualized by Silver Stain Plus (Bio-Rad).
Acquisition of image and data analysis
The gel images were scanned using the Fluor-S MultiImager system and analyzed with PDQuest version 7.2.0 (Bio-Rad). To compare spots across gels, a standard image of three biological replicates was created for each sample. The quantity and quality scores to each spot were assigned as described56. The correlation coefficient, representing the association between the spot intensities on replicates, was maintained at a minimum of 0.8. The spot densities on the “standard gel” were normalized against the total density in the gel image. To facilitate the comparison of the “standard gels”, the spot volumes were further normalized using three unaltered protein spots across all of the gels. The filtered spot quantities from the “standard gels” were assembled into a data matrix for further analysis.
Analysis of the 5′-upstream region of CaTLPs
Genome-walking PCR was performed using the Genome Walker kit (Clontech Laboratories). Primary and secondary PCR were conducted using adaptor primers, and adaptor and nested primers complementary to the CaTLP1 upstream sequence (Supplemental Table 5). The amplicon was cloned into pENTR/D-TOPO vector (Invitrogen). The promoter region of CaTLP2 and CaTLP3 were retrieved from the Next Generation Challenge Programme on Chickpea (NGCPC) facility at National Institute of Plant Genome Research, New Delhi, India. Sequence analysis was done using PLACE (http://www.dna.affrc.go.jp/htdocs/PLACE/signalscan.html) and PlantCARE (http://bioinformatics.psb.gent.be/webtools/plantcare/html/) databases.
Histochemical detection of GUS
GUS histochemical analysis was performed58 with tissues or whole seedlings submerged in GUS staining buffer [2 mM 5-bromo-4-chloro-3-indolyl glucuronide, 0.1 M sodium phosphate (pH 7.0), 0.1% (v/v) Triton X-100, 1 mM potassium ferricyanide, 1 mM potassium ferrocyanide and 10 mM EDTA], vacuum infiltrated for 10 min and incubated overnight at 37 °C. The samples were treated with 95% (v/v) ethanol and observed with a stereomicroscope (SZX-ILLD2-200, Olympus).
Immunoprecipitation
Immunoprecipitation was performed with rProtein A Sepharose Fast Flow (GE Healthcare). Protein extracts were prepared by homogenization of 0.5 g tissue each from wild-type and CaTLP1-FLAG overexpressing Arabidopsis lines in 2 mL buffer containing 50 mM HEPES-KOH, pH 7.5, 0.15 M NaCl, 0.5% (v/v) Triton X-100, and 0.1% (v/v) Tween 20. The homogenates were centrifuged at 10,000 g for 15 min at 4 °C to remove cellular debris. The supernatants were incubated with sepharose beads at 4 °C for 16 h to remove non-specific proteins. Beads were separately incubated with anti-FLAG antibody (Agrisera) at 23 ± 2 °C for 1 h. The pre-extracts were then incubated with anti-FLAG conjugated beads at 4 °C for 16 h, and the beads with immunoaffinity complexes were retrieved. The immunoaffinity complexes were eluted with 50 mL of 2x SDS-sample buffer containing 100 mM Tris-HCl, pH 6.8, 4% (w/v) SDS, 20% (w/v) glycerol and 5% (v/v) 2-mercaptoethanol.
Immunoblot analysis
Immunoblot analysis was performed by resolving proteins on 12.5% SDS-PAGE and electrotransferring onto nitrocellulose membrane at 150 mA for 2 h. The membranes were blocked with 5% (w/v) non-fat milk for 1 h, and incubated with appropriate antibodies for 2 h. The blots were then incubated with alkaline phosphatase conjugated secondary antibody for 1 h and the signals were detected using NBT/BCIP (nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate) method.
Mass spectrometry analysis
The immunoprecipitated proteins were subjected to trypsinolysis as described56. The peptides were analyzed using a QSTAR Elite mass spectrophotometer (Applied Biosystem) coupled with an on-line Tempo nano-MDLC system. Proteins were identified by the Mascot search algorithm56. The function of proteins was assigned using a protein function database Pfam (http://www.sanger.ac.uk/software/Pfam/) or Inter-Pro (http://www.ebi.ac. uk/interpro/).
In silico protein interaction analysis
The interactions of CaTLP1, CaTLP2, casein kinase 1 like 2 and CaCK1 were carried out in the global network using homology modeling. Each of the candidate proteins was searched for their corresponding template via PDB-BLAST. Interaction of CaTLP1 and CaTLP2 was performed with proteins identified in immunoprecipitated complexes from Arabidopsis and chickpea using standalone ZDOCK version 3.0.2. NetPhosK 1.0 server (http://www.cbs.dtu.dk/services/NetPhosK/) was used to predict the probable sites of phosphorylation and the kinase-specific interactome was constructed in NetworKIN (version 2.0)59. Visualization and analysis of interaction networks were accomplished on Cytoscape version 2.8.1.
BiFC assay
Coding sequences were cloned in fusion with the N- and C-terminal parts of YFP under the control of the CaMV35S promoter in the BiFC vectors. The fluorescence complementation was assayed by transient expression using all eight compatible combinations between protein pairs. Each expression vector was introduced in Agrobacterium tumefaciens strain GV 3101 by electroporation. The bacterial cultures were incubated overnight at 28 °C. Each culture was pelleted, washed, and resuspended in infiltration buffer [for 50 ml buffer 250 mg D-glucose, 5 ml of 500 mM MES, 5 ml of 20 mM Na3PO4.12H2O 5 μl of 1 M acetosyringone] to an OD600 of 0.5. The inoculum was delivered to the lamina tissue of N. benthamiana leaves by infiltration through the lower epidermis. To enhance transient expression of BiFC fusion proteins, the P19 viral suppressor of gene silencing was coexpressed60. YFP fluorescence was detected 2 to 3 d after infiltration.
Statistical analysis
All the experiments were performed in triplicate. The experimental results were expressed as the mean ± standard deviation (SD) using SPSS (Statistical Program for Social Sciences, SPSS Corporation, Chicago, Illinois, USA). Further, one-way analysis of variance (ANOVA) and post hoc analysis with 2-sided Tukey’s HSD were performed at p ≤ 0. 05.
Additional Information
How to cite this article: Wardhan, V. et al. Chickpea transcription factor CaTLP1 interacts with protein kinases, modulates ROS accumulation and promotes ABA-mediated stomatal closure. Sci. Rep. 6, 38121; doi: 10.1038/srep38121 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Leung, J. & Giraudat, J. Abscisic acid signal transduction. Annu. Rev. Plant Physiol. Plant Mol. Biol. 49, 199–222 (1998).
Himmelbach, A., Iten, M. & Grill, E. Signalling of abscisic acid to regulate plant growth. Philos. Trans. R. Soc. L. B. Biol. Sci. 353, 1439–1444 (1998).
Ingram, J. & Bartels, D. The molecular basis of dehydration tolerance in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 377–403 (1996).
Shinozaki, K. & Yamaguchi-Shinozaki, K. Gene Expression and Signal Transduction in Water-Stress Response. Plant Physiol. 115, 327–334 (1997).
Shinozaki, K., Yamaguchi-Shinozaki, K. & Seki, M. Regulatory network of gene expression in the drought and cold stress responses. Curr. Opin. Plant Biol. 6, 410–7 (2003).
Yamaguchi-Shinozaki, K. & Shinozaki, K. Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci. 10, 88–94 (2005).
Noben-Trauth, K., Naggert, J. K., North, M. A. & Nishina, P. M. A candidate gene for the mouse mutation tubby. Nature 380, 534–538 (1996).
Mukhopadhyay, S. & Jackson, P. K. The tubby family proteins. Eye 6, 3 (2011).
Lai, C.-P. et al. Molecular analyses of the Arabidopsis TUBBY-like protein gene family. Plant Physiol. 134, 1586–1597 (2004).
Ikeda, S. et al. Cell-specific expression of tubby gene family members (tub, Tulp1,2, and 3) in the retina. Invest. Ophthalmol. Vis. Sci. 40, 2706–2712 (1999).
Kou, Y., Qiu, D., Wang, L., Li, X. & Wang, S. Molecular analyses of the rice tubby-like protein gene family and their response to bacterial infection. Plant Cell Rep. 28, 113–121 (2009).
Yang, Z. et al. Genomewide comparative phylogenetic and molecular evolutionary analysis of tubby-like protein family in Arabidopsis, rice, and poplar. Genomics 92, 246–253 (2008).
Reitz, M. U. et al. The subcellular localization of Tubby-like proteins and participation in stress signaling and root colonization by the mutualist Piriformospora indica. Plant Physiol. 160, 349–364 (2012).
Bhushan, D. et al. Comparative proteomics analysis of differentially expressed proteins in chickpea extracellular matrix during dehydration stress. Mol. Cell. Proteomics 6, 1868–1884 (2007).
Wardhan, V. et al. Overexpression of CaTLP1, a putative transcription factor in chickpea (Cicer arietinum L.), promotes stress tolerance. Plant Mol. Biol. 79, 479–493 (2012).
Singh, N. D., Li, M., Lee, S.-B., Schnell, D. & Daniell, H. Arabidopsis Tic40 expression in tobacco chloroplasts results in massive proliferation of the inner envelope membrane and upregulation of associated proteins. Plant Cell 20, 3405–3417 (2008).
Tamura, S. et al. Bioisostere of valtrate, anti-HIV principle by inhibition for nuclear export of Rev. Bioorg. Med. Chem. Lett. 20, 2159–2162 (2010).
Ito, T. et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 98, 4569–74 (2001).
Neubauer, G. et al. Identification of the proteins of the yeast U1 small nuclear ribonucleoprotein complex by mass spectrometry. Proc. Natl. Acad. Sci. USA 94, 385–90 (1997).
Gavin, A.-C., Maeda, K. & Kuhner, S. Recent advances in charting protein-protein interaction: mass spectrometry-based approaches. Curr. Opin. Biotechnol. 22, 42–49 (2011).
Moradas-Ferreira, P., Costa, V., Piper, P. & Mager, W. The molecular defences against reactive oxygen species in yeast. Mol. Microbiol. 19, 651–658 (1996).
Westgate, M. E. & Boyer, J. S. Osmotic adjustment and the inhibition of leaf, root, stem and silk growth at low water potentials in maize. Planta 164, 540–549 (1985).
Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246 (2013).
Hatakeyama, S. et al. Ubiquitin-dependent degradation of IκBα is mediated by a ubiquitin ligase Skp1/Cul 1/F-box protein FWD1. Proc. Natl. Acad. Sci. USA 96, 3859–3863 (1999).
Grima, B. et al. The F-box protein Slimb controls the levels of clock proteins period and timeless. Nature 420, 178–182 (2002).
Hu, C. D., Chinenov, Y. & Kerppola, T. K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798 (2002).
Chinnusamy, V., Jagendorf, A. & Zhu, J. K. Understanding and improving salt tolerance in plants. Crop Sci. 45, 437 (2005).
Kamada, Y., Jung, U. S., Piotrowski, J. & Levin, D. E. The protein kinase C-activated MAP kinase pathway of Saccharomyces cerevisiae mediates a novel aspect of the heat shock response. Genes Dev. 9, 1559–1571 (1995).
Flattery-O’Brien, J., Collinson, L. P. & Dawes, I. W. Saccharomyces cerevisiae has an inducible response to menadione which differs from that to hydrogen peroxide. J. Gen. Microbiol. 139, 501–507 (1993).
Schnell, N., Krems, B. & Entian, K. D. The PAR1 (YAP1/SNQ3) gene of Saccharomyces cerevisiae, ac-jun homologue, is involved in oxygen metabolism. Curr. Genet. 21, 269–273 (1992).
Stephen, D. W., Rivers, S. L. & Jamieson, D. J. The role of the YAP1 and YAP2 genes in the regulation of the adaptive oxidative stress responses of Saccharomyces cerevisiae. Mol. Microbiol. 16, 415–423 (1995).
Krems, B., Charizanis, C. & Entian, K. D. Mutants of Saccharomyces cerevisiae sensitive to oxidative and osmotic stress. Curr. Genet. 27, 427–434 (1995).
Kuge, S. & Jones, N. YAP1 dependent activation of TRX2 is essential for the response of Saccharomyces cerevisiae to oxidative stress by hydroperoxides. EMBO J. 13, 655–664 (1994).
Tuberosa, R. et al. Identification of QTLs for root characteristics in maize grown in hydroponics and analysis of their overlap with QTLs for grain yield in the field at two water regimes. Plant Mol. Biol. 48, 697–712 (2002).
Steppuhn, H. & Raney, J. P. Emergence, height, and yield of canola and barley grown in saline root zones. Can. J. Plant Sci. 85, 815–827 (2005).
Leckie, C. P., McAinsh, M. R., Allen, G. J., Sanders, D. & Hetherington, A. M. Abscisic acid-induced stomatal closure mediated by cyclic ADP-ribose. Proc. Natl. Acad. Sci. USA 95, 15837–42 (1998).
Schroeder, J. I., Kwak, J. M. & Allen, G. J. Guard cell abscisic acid signalling and engineering drought hardiness in plants. Nature 410, 327–330 (2001).
Oono, Y. et al. Monitoring expression profiles of Arabidopsis gene expression during rehydration process after dehydration using ca 7000 full-length cDNA microarray. Plant J. 34, 868–887 (2003).
Kant, P. et al. Functional-genomics-based identification of genes that regulate Arabidopsis responses to multiple abiotic stresses. Plant Cell Environ. 31, 697–714 (2008).
Magnan, F. et al. Mutations in AtCML9, a calmodulin-like protein from Arabidopsis thaliana, alter plant responses to abiotic stress and abscisic acid. Plant J. 56, 575–589 (2008).
Zolla, L., Rinalducci, S., Antonioli, P. & Righetti, P. G. Proteomics as a complementary tool for identifying unintended side effects occurring in transgenic maize seeds as a result of genetic modifications. J. Proteome Res. 7, 1850–1861 (2008).
Chakraborty, N. et al. Reduction of oxalate levels in tomato fruit and consequent metabolic remodeling following overexpression of a fungal oxalate decarboxylase. Plant Physiol. 162, 364–378 (2013).
Corpillo, D., Gardini, G., Vaira, A. & Basso, M. Proteomics as a tool to improve investigation of substantial equivalence in genetically modified organisms: The case of a virus‐resistant tomato. Proteomics 4, 193–200 (2004).
Lehesranta, S. J. et al. Comparison of tuber proteomes of potato varieties, landraces, and genetically modified lines. Plant Physiol. 138, 1690–1699 (2005).
Agrawal, L. et al. Comparative proteomics reveals a role for seed storage protein AmA1 in cellular growth, development, and nutrient accumulation. J. Proteome Res. 12, 4904–4930 (2013).
Giraudat, J. et al. Current advances in abscisic acid action and signalling. Plant Mol. Biol. 26, 1557–1577 (1994).
Busk, P. K. & Pages, M. Regulation of abscisic acid-induced transcription. Plant Mol. Biol. 37, 425–435 (1998).
Pawson, T. & Nash, P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 14, 1027–47 (2000).
Choi, H., Hong, J., Ha, J., Kang, J. & Kim, S. Y. ABFs, a family of ABA-responsive element binding factors. J. Biol. Chem. 275, 1723–1730 (2000).
Chang, Y. F. & Carman, G. M. Casein kinase II phosphorylation of the yeast phospholipid synthesis transcription factor Opi1p. J. Biol. Chem. 281, 4754–4761 (2006).
Plotnikov, A., Chuderland, D., Karamansha, Y., Livnah, O. & Seger, R. Nuclear extracellular signal-regulated kinase 1 and 2 translocation is mediated by casein kinase 2 and accelerated by autophosphorylation. Mol. Cell. Biol. 31, 3515–3530 (2011).
Yoshioka, K. et al. The chimeric Arabidopsis Cyclic Nucleotide-Gated Ion Channel 11/12 activates multiple pathogen resistance responses. Plant Cell 18, 747–763 (2006).
Silva, H., Yoshioka, K., Dooner, H. K. & Klessig, D. F. Characterization of a new Arabidopsis mutant exhibiting enhanced disease resistance. Mol. Plant Microbe Interact. 12, 1053–1063 (1999).
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Lehmann, T. & Pollmann, S. Gene expression and characterization of a stress-induced tyrosine decarboxylase from Arabidopsis thaliana. FEBS Lett. 583, 1895–1900 (2009).
Subba, P. et al. Phosphoproteomic dynamics of chickpea (Cicer arietinum L.) reveals shared and distinct components of dehydration response. J Proteome Res. 12, 5025–5047 (2013).
Agrawal, L. et al. Comparative proteomics of tuber induction, development and maturation reveal the complexity of tuberization process in potato (Solanum tuberosum L.). J Proteome Res. 7, 3803–3817 (2008).
Jefferson, R. A., Kavanagh, T. A. & Bevan, M. W. GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6, 3901–3907 (1987).
Linding, R. et al. Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426 (2007).
Canto, T., Uhrig, J. F., Swanson, M., Wright, K. M. & MacFarlane, S. A. Translocation of Tomato Bushy Stunt Virus P19 protein into the nucleus by ALY proteins compromises its silencing suppressor activity. J Virology, 80, 9064–9072 (2006).
Acknowledgements
This work was supported by grants (BT/PR13045/PBD/16/863/2009) from the Department of Biotechnology (DBT), Government of India to N.C. The authors thank Council of Scientific and Industrial Research (CSIR), Government of India for providing pre-doctoral fellowship to V.W. and the National Institute of Plant Genome Research, New Delhi for providing post-doctoral fellowship to A.P. We kindly acknowledge Dr. T. Nakagawa, Shimane University, Japan for the generous gift of destination vectors pGWB411, 433 and 441. We express our gratitude to Dr. David Bouchez, Institut Jean-Pierre Bourgin, Versailles, France for complete set of four pBiFP vectors.
Author information
Authors and Affiliations
Contributions
N.C. conceived the project. V.W., S.C. and N.C. designed the study. V.W. performed the experiments. V.W., S.C and N.C. analyzed the data and wrote the article. A.P. contributed to writing the article.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wardhan, V., Pandey, A., Chakraborty, S. et al. Chickpea transcription factor CaTLP1 interacts with protein kinases, modulates ROS accumulation and promotes ABA-mediated stomatal closure. Sci Rep 6, 38121 (2016). https://doi.org/10.1038/srep38121
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38121
This article is cited by
-
Overexpression of a rice Tubby–like protein-encoding gene, OsFBT4, confers tolerance to abiotic stresses
Protoplasma (2023)
-
AtTLP2, a Tubby-like protein, plays intricate roles in abiotic stress signalling
Plant Cell Reports (2022)
-
Tubby-like proteins (TLPs) transcription factor in different regulatory mechanism in plants: a review
Plant Molecular Biology (2022)
-
A tubby-like protein CsTLP8 acts in the ABA signaling pathway and negatively regulates osmotic stresses tolerance during seed germination
BMC Plant Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
