
Abstract
Since the sequencing of large genomes, many statistical features of their sequences have been found. One intriguing feature is that certain subsequences are much more abundant than others. In fact, abundances of subsequences of a given length are distributed with a scale-free power-law tail, resembling properties of human texts, such as Zipf’s law. Despite recent efforts, the understanding of this phenomenon is still lacking. Here we find that selfish DNA elements, such as those belonging to the Alu family of repeats, dominate the power-law tail. Interestingly, for the Alu elements the power-law exponent increases with the length of the considered subsequences. Motivated by these observations, we develop a model of selfish DNA expansion. The predictions of this model qualitatively and quantitatively agree with the empirical observations. This allows us to estimate parameters for the process of selfish DNA spreading in a genome during its evolution. The obtained results shed light on how evolution of selfish DNA elements shapes non-trivial statistical properties of genomes.
Similar content being viewed by others

Introduction
Our genome is a sequence of A, C, G and T nucleotides and can be viewed as a long text of about three billion letters. Only a small part of our genome is functional and under selection1,2,3; the rest (so-called junk DNA) mostly evolves neutrally and, therefore, is naively expected to be a random sequence. However, the junk DNA contains many homologous sequences, sharing significant similarities to each other. Hence, its statistical properties differ from those of random sequences4,5,6,7. One of these properties, which we discuss here, is that for a given length, certain subsequences are much more abundant than others8,9,10,11. Namely, the abundances of k-mers—sequences of length k—possess a wide, scale-free distribution, as shown in Fig. 1. One can see that even for large values of k, one finds k-mers which appear more than 104 times in the human genome, while in a randomly shuffled genome such k-mers would be unique.

Distributions of abundances of k-mers for k = 40.
s is the number of copies of a certain k-mer and nk(s) is the number of different k-mers with abundance s. Distributions for different genomic compartments are presented: the whole genome (solid, black), the whole genome after masking the repeat elements (solid, green) and the Alu family of repeats (solid, blue). For comparison the distribution of word abundances in Pride and Prejudice72 is also shown (solid, red). The dashed line represents the power-law nk(s) ~ s−α with α = 2. For a randomly shuffled human genome or a random sequence of the same length there is not a single k-mer with s > 1. Inset: the corresponding Zipf’s plots for the main figure. For each k-mer (or a word for Pride and Prejudice) its abundance is plotted vs. the rank of its abundance. The dashed line represents the power-law s ~ 1/rank(s).
This phenomenon resembles statistical properties of human texts, where abundances of words also exhibit a scale-free distribution12. For human texts such a linguistic feature is often presented as Zipf’s13,14 or Heaps’15 law. We exemplify the similarity between the statistics of k-mers in the human genome and the statistics of words in human texts in Fig. 1. Despite an incomplete analogy, caused by the lack of a natural definition of a word in the genomic context, this intriguing similarity between the genome and human texts has led some researchers to analyze genetic sequences from a linguistic perspective (see, e.g., refs 16,17). However, beyond human texts, many types of data in the physical and social sciences can be approximated with a Zipf distribution, so that the observation that genomic texts obey Zipf’s law is not enough to prove genomes have linguistic features. Linguistic features include morphology, syntax and semantics and Zipf’s distribution alone is not specific to language. This is why several studies called linguistic properties of genomes into question18,19,20,21,22,23,24,25. Here we present an explanation for the observed genomic phenomenon, showing that the pseudo-linguistic features of the k-mer abundances statistics in genomes are a consequence of selfish DNA expansion in our genome during its evolution. We develop a model, which accurately reproduces statistical properties of abundant subsequences in the genome. The model is based solely on selfish spreading of DNA repeats, demonstrating that high abundances of certain k-mers do not reflect their functionality for the organism.
Considering different compartments of the genome, one finds that the scale-free distribution of abundances is dominated by subsequences of selfish repetitive DNA elements (see Fig. 1). This suggests that the scale-free distribution of abundances is a consequence of the evolutionary dynamics of such elements. Selfish DNA elements (or repeats) are parasitic sequences that duplicate with the help of the cellular machinery of the host organism26,27. Such duplications significantly increase the size of genomes during their evolution and often appear in bursts of activity during a few tens of millions of years28,29. After such a burst, the duplication activity stops, but the existing repetitive elements remain in the genome. Some elements acquire a function30 or cause a disease31, but most fade away neutrally into the genomic background due to mutations32. One of the largest and most studied families of repeats in primates is the Alu family, covering 15% of the human genome with more than a million copies33,34. In the following, we use the Alu family as a model system to study statistical properties of selfish DNA sequences.
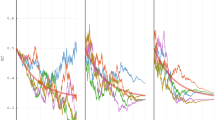
To gain insight into the origin of the observed fat-tailed distributions of k-mer abundances in the human genome, we plot in Fig. 2 the distributions for the Alu family elements for different values of k (see Methods for details). One can see that even the abundances of short k-mers are much more dispersed than in a random sequence. For large values of k > 20 the distributions possess a power-law tail, i.e. nk(s) ~ s−α. Importantly, the exponent of the power-law distribution, α(k), depends on k, such that it increases with k, starting from about 2 for small values of k. This dependence is clearly visible in Fig. 3, where we measure the values of α(k) using the Hill estimator (see Methods). A model for the evolutionary dynamics of selfish DNA ought to be able, in particular, to explain these properties of the power-law exponent and, in general, to reproduce the empirical distributions.

Distributions of abundances of k-mers, nk(s), for different values of k, from 5 to 90 in steps of 5, from top to bottom (see numbers in the figure).
Circles represent nk(s) in the empirical data for the Alu family of repeats (see Methods). Dots represent nk(s) in a random sequence, of the same length as the empirical one for k = 5 (red) and k = 10 (blue). Lines represent nk(s) in simulated Alu elements using the set of parameters in Eq. (9) in Methods. The results of the simulations do not change drastically, as long as the parameters are within the ranges specified in Eq. (7). The dashed lines represent the power-law decay ns ~ s−α with α = 2. For visibility the values of nk(s) are normalized differently for each value of k (but in the same way for the empirical and the simulated data), so that the units of the vertical axis are arbitrary.

Estimation of parameters of the model using the analytic fit of the empirical data.
Circles represent the empirical power-law exponent α as a function of k. The line is the numerical fit of the data points using Eq. (3), resulting in Eq. (4). The analytic equation for the fit and the resulting estimator are presented in the upper-left corner. For details of the estimators and the fit see Methods.
In this paper we present a simple model for the evolution of selfish DNA, which accounts qualitatively and quantitatively for the observed distributions of k-mer abundances. Using our model, we estimate key parameters of the spreading dynamics of Alu repeat elements and compare them to previous estimates. Our results demonstrate that some non-trivial properties of genomic texts can be understood considering the evolution of selfish DNA, without referring to any linguistic structure of genomes.
The model
We analyze a model for the evolution of selfish DNA in a genome, which is based on the assumption that every repeat element can be active and duplicate with a certain rate or passive and does not duplicate at all. In a sense our model is a mixture between the transposon model, which assumes that every Alu element duplicates with the same rate35 and the master gene model, which assumes that there is a single active, duplication potent element which gives rise to all other silent elements36,37. Our approach of mixing the two models is supported by recent studies, which suggest that the fraction of active Alu elements is not 100% as in the transposon model nor extremely small as in the master gene model38,39,40.
In our model the process starts from the appearance of a single active (i.e. able to duplicate) selfish element of length L at time t = 0. During the burst of activity, in the time interval 0 ≤ t ≤ T1, all existing active elements duplicate in a genome with rate γ. Each duplication results in a new identical element, which we assume to be active with probability δ and silent (non-duplicating) with probability 1 − δ. This results in an exponential growth, such that the average number of elements after the burst of activity ends at time t = T1 is given by

During such a burst, these duplications shape a branching process, which gives rise to a phylogenetic tree, as illustrated in Fig. 4. After the burst, the duplication activity is suppressed and all N elements are silenced for a time period T2. We observe these elements in the present day genome, at time t = T1 + T2. During and after the burst, all existing elements accumulate mutations with rate μ0 per bp, except CpG nucleotides, which mutate approximately 6 times faster41. We define the effective mutation rate μ as the weighted average of the two rates. An illustration of the described model for the evolution of selfish DNA elements is presented in Fig. 4.

Illustration of the analysed model for the dynamics of repeat elements. Each branch represents a sequence of the repeat.
Active elements are depicted in thick, black lines, while silent ones are shown in thin, gray lines. During the activity burst, selfish elements duplicate exponentially with time and accumulate mutations (red marks). After the burst sequences do not duplicate anymore but still mutate.
To address the empirically observed scale-free distribution of k-mer abundances in genomic data, we consider in particular the statistics of k-mers in this model. There are L − k + 1 k-mers in a single element. A duplication event increases the number of all k-mers in the duplicating element, while mutations decrease abundances of certain k-mers and increase abundances of others. The mutation rate of a k-mer is μk, such that the probability that a k-mer does not mutate for a time T2 is given by  .
.
Using two simplifying assumptions, we solve the model analytically (see detailed derivation in the SI). The analytic solution of the model yields that the number of k-mers with abundance  at present time t = T1 + T2, which we denote by nk(s), is given by
at present time t = T1 + T2, which we denote by nk(s), is given by

Here N is the number of repeat elements at present time, given by Eq. (1). The power-law exponent of the distribution is

and  is the probability of a k-mer to preserve its sequence during the second, silent phase.
is the probability of a k-mer to preserve its sequence during the second, silent phase.
Note, that the power-law tail exists only if, on average, a k-mer duplicates faster than it mutates, such that μk < δγ. In the context of this paper this condition is fulfilled. The derived dependence for the power-law exponent α in Eq. (3) accounts for the observations presented above: α(k) is predicted to increase with k, starting from α(k) = 2 for small values of k.
To further quantitatively test the presented model, one needs to estimate the parameters N (number of elements), μ (effective mutation rate), T1 (time of the first, active phase), T2 (time of the second, silent phase) and δ (probability of a new element to be active). The duplication rate, γ can be then estimated using Eq. (1). We obtain the estimates using the empirical data and the analytic result (3). As we show below, our model accurately reproduces the empirical data for the Alu family of repeats for the estimated set of parameters.
Modeling evolution of Alu repeats
The presented model can be used to study the evolution of large selfish DNA families. We apply it here to the Alu family of repeats, studying distributions of k-mer abundances in all identified Alu repeats in the human genome, excluding the still active AluY subfamily42 (see Methods for more details). In Fig. 2 one can see that these distributions qualitatively agree with the predictions of Eqs (2) and (3): the tails of the distributions follow a power-law, the exponents of these power-laws are larger than 2 and grow with k (see also Fig. 3).
We start now with the estimation of the parameters of the model. First, we estimate the ratio  using the analytic result (3). From the empirical data the value of the power-law exponent α(k) can be estimated for each k using the Hill maximum-likelihood estimator43 (see Methods). In Fig. 3 one can see the agreement of Eq. (3) with the empirical power-law exponents, using a fit with a single free parameter,
using the analytic result (3). From the empirical data the value of the power-law exponent α(k) can be estimated for each k using the Hill maximum-likelihood estimator43 (see Methods). In Fig. 3 one can see the agreement of Eq. (3) with the empirical power-law exponents, using a fit with a single free parameter,  . The fitting results in
. The fitting results in

To estimate the effective mutation rate, μ, it is important to consider hypermutable CpG di-nucleotides along the Alu elements. In fact, μ is the average mutation rate of CpG di-nucleotides and other nucleotides. There are 24 CpG di-nucleotides in a typical Alu element (e.g. in the consensus sequence of the AluSx subfamily) of length L ≃ 300. These di-nucleotides mutate about 6 times faster than other nucleotides on both positions41. Thus,

where μ0 is the mutation rate of the non-CpG nucleotides and μCpG = 6μ0 is the mutation rate of the CpG nucleotides. In the following we measure all the rates in units of μ0, which is of the order of 10−9 yr−1 and times in units of  . The value of μ0 is just a global time scale and does not affect the k-mer abundances. Nevertheless, in the Discussion section we estimate μ0, convert all estimated parameters to standard units and compare them with previous estimates in the literature.
. The value of μ0 is just a global time scale and does not affect the k-mer abundances. Nevertheless, in the Discussion section we estimate μ0, convert all estimated parameters to standard units and compare them with previous estimates in the literature.
The probability of a new element to be active, δ, does not affect the results in the asymptotic limit of large numbers of Alu elements, N, as long as the effective duplication rate, δγ, is kept constant. However, for a finite value of N the results change if δ is too small. In this case estimates of α(k) would be biased to higher values due to highly abundant copies of several active elements, such that Eq. (3) would not fit well the biased estimates. The fact that Eq. (3) does fit well the empirical data indicates that δ is not very small. Our simulations, with N = 776710 and Eq. (4), indicate that the distribution of abundances does not depend significantly on δ and Eq. (3) fits well the data, as long as δ is above 5%. This result supports an earlier study, where δ is estimated to be 10–20%44. Using our estimate δ = (5–100)% and Eqs (4) and (5) we conclude that the duplication rate is in the range γ = (0.2–4) ⋅ 103μ0. Furthermore, using the above estimates together with Eq. (1) we get the estimate T1 = (5.3–6.9) ⋅ 10−2/μ0.
To find the only remaining missing parameter T2, we use the independence of the results on the value of δ in the relevant regime, setting δ = 1 and simulating the model for many different values of T2. The best agreement between the empirical distribution of abundances and the simulated one was obtained for

More details about the estimation of the parameters from the empirical data can be found in Methods.
In summary, the estimated parameter set for the Alu family evolution model is

As shown in Fig. 2, the model with this set of parameters accurately reproduces the empirical distributions of the k-mer abundances for the Alu elements.
Discussion
There are a few important things to note before we draw conclusions and summarize. First, the presented model is similar in spirit to the one suggested in ref. 10. However, the basic assumption there was that the evolution of the selfish DNA elements approaches a steady state with a constant genome size, such that any new element replaces an old one, resulting in  . As has been shown in ref. 10, this assumption can only result in an abundance distribution following
. As has been shown in ref. 10, this assumption can only result in an abundance distribution following  , such that α = 1 for all values of k. In contrast, we assume an exponentially growing steady state of the genome in the burst phase,
, such that α = 1 for all values of k. In contrast, we assume an exponentially growing steady state of the genome in the burst phase,  (see SI). The last assumption makes more biological sense for the expansion of selfish DNA, with a weak or no selection against it. Only in this case, when there is a phase of exponentially expanding repeats, one can get a power-law exponent α(k) which is always larger than 2 and depends on k, as it is observed for the empirical data.
(see SI). The last assumption makes more biological sense for the expansion of selfish DNA, with a weak or no selection against it. Only in this case, when there is a phase of exponentially expanding repeats, one can get a power-law exponent α(k) which is always larger than 2 and depends on k, as it is observed for the empirical data.
In our model we assumed that CpG di-nucleotides mutate 6 times faster than other nucleotides. This assumption results in an effective mutation rate of Alu elements which is 1.8 times higher than the mutation rate of non-CpG nucleotides elsewhere in the genome (see Eq. (5)). A tempting simplification of the model would be to ignore the CpG di-nucleotides, assuming an effective mutation rate for all nucleotides along an Alu element. However, in that case, the distribution obtained for k-mer abundances is qualitatively different from the empirical distribution (see SI for more details). Here, we only stress that modeling non-uniform mutation rate with highly mutable CpG di-nucleotides is essential to account for the empirical data.
Evolution of repeat elements in our genome is a complex process, which probably involves selection, population dynamics and other factors28,45,46,47,48. Detailed studies of Alu repeats reveal a complex history with many subfamilies appearing at different times29,49,50,51,52,53,54. As it often happens in nature, very complex phenomena tend to exhibit random-like statistical features. For instance, complex speciation processes result in a simple Yule statistics of genera sizes55 and simple statistics of pairwise genomic distances56; complex biochemical processes result, to some extent, in simple molecular clocks with effective mutation rates of nucleotides and amino acids on the evolutionary timescale57,58, etc. This study suggests another example of this kind: a complex evolution of selfish DNA elements exhibits random-like properties with some effective parameters.
Our estimates of those effective parameters might suffer from various biases. The first stems from the fact that we assumed a constant mutation rate along the human lineage since the origin of the Alu family in the genome, which might have varied, for instance due to different generation times59,60. Moreover, Alu elements are enriched (relative to the whole genome) with CpG di-nucleotides which possess an order of magnitude higher mutation rate41,61,62. We assumed that the mutation rate of the CpG di-nucleotides is 6 times higher than that of other nucleotides, but in reality the situation might be more complex41. Positive or negative selection can increase or decrease the estimate for the effective mutation rate. The duplication rate can possess more complex temporal structure than the one we assumed in our model and may depend on the sequence of the element29. These and other effects are, probably, the reason for the disagreement between our simulations and the empirical distributions at small abundances and, for very long k-mers, at the very end of their tails (see Fig. 2). Another possible reason for this disagreement is an intrinsic stochasticity of the simulations. Performing other simulations with a different random seed, one finds that the simulations disagree in the same regions where the simulation disagrees with the empirical results (see Fig. 2 and Fig. S2 in SI). This means that in those regions using our stochastic model one cannot fit the empirical results because the results of the simulations significantly depend upon the realization of the model (random seed). In other regions, however, the results of the simulations are very robust to the stochastic noise and agree well with the empirical results.
Our estimate for the age of the Alu family in units of the neutral mutation rate is T1 + T2 = (7.7–9.3) ⋅ 10−2/μ0. Alternatively, one can estimate the age of the Alu family from the following phylogenetic arguments. The Alu family is primate specific63, so we expect that the age of Alu is about 80 ⋅ 106 yr64. Therefore, our estimate for the neutral mutation rate turns out to be about μ0 = (0.9–1.1) ⋅ 10−9 yr−1, within the range estimated in the literature60. Furthermore, in this case our estimate of T2 = (20–24) ⋅ 106 yr is in a rough agreement with the conclusion of ref. 65 that “most human Alu sequences were inserted in a process that ceased about 30 million years ago”. A similar estimate was obtained in ref. 36. Therefore, our estimates of the parameters yield reasonable values, suggesting that the above discussed possible biases are not of great importance in the context of this study.
In the literature there are two alternative models for the expansion of Alu elements. The first one is the transposon model, which assumes that every Alu element duplicates with the same rate35. This corresponds to δ = 1 in our model. The second one, the master gene model, assumes δ = 0, implying that there is a single active, duplication potent element which gives rise to all other elements36,37. More recent studies suggest that the fraction of active Alu elements, δ, is not 100% as in the transposon model nor extremely small as in the master gene model38,39,40. This fraction for the Alu family was estimated to be 10–20%44. From our simulations we find that Eq. (3) is expected to fit the data well as long as δ is larger than 5%. Therefore, the fact that the empirical data is well fitted by Eq. (3) (see Fig. 3) supports the estimate in ref. 44.
Since the assumptions of our model are quite general, it is expected to capture the dynamics of evolution of many selfish elements in many species. Indeed, abundance distributions of k-mers of the Alu elements in the chimp genome are very similar to the ones in the human genome (see Fig. S3 in SI). Beyond the Alu family, several large repeat families in other species distant from human demonstrate the behaviour predicted by our model: the k-mer abundance distributions possess a power-law tail, its power-law exponent is above 2 and grows with k, following Eq. (3) (see Figs S4–S7 in SI).
Selfish elements cover a significant fraction of our genome, resulting in a genome-wide power-law distribution of k-mer abundances. Since different selfish DNA families evolve with different effective parameters, their mixture—the genome-wide power-law—is not expected to be clean. However, the main predicted feature of our model is that the power-law exponent has to be larger than and close to 2, as it is observed. Thus, our results explain the genome-wide power-law in th k-mer abundance distributions and, therefore, in the Zipf plot of k-mers in genomes, without referring to any linguistic or functional features. In fact, we demonstrate that a high abundance of a certain subsequence is not necessarily due to its functionality for an organism, but may rather reflect its ability to parasitize and selfishly spread in the host genome. The presented simple model of selfish DNA evolution surprisingly accurately accounts for statistical properties of these highly abundant subsequences in our genome.
Methods
Data
The sequences of all identified Alu repeat elements in the human genome were downloaded from the Ensembl database using the Perl API66. We filter out the X and Y chromosomes and the sequences belonging to the still active AluY subfamily42. In Fig. 1 the AluY subfamily is not filtered out. Sequences of other repeat elements in other species were downloaded using the table browser from the UCSC Table Browser data retrieval tool67. The k-mer abundances were counted using the Jellyfish program68. To smooth the resulting distribution of abundances in Fig. 2, we used logarithmic binning for s > 7, such that the ratio between two neighbouring values of s is 1.1885.
Simulations
First we computed a branching pattern (or phylogenetic tree) of selfish elements as shown schematically in Fig. 4. This branching process was simulated in continuous time using a Kinetic Monte Carlo scheme69. We start with one active element at t = 0. At any given point in time all possible future branchings of active elements are listed; each of them occur with rate γ. Drawing a random number one of them is selected and executed; the time then advanced appropriately. The new element is active, i.e. able to duplicate again, with probability δ. Drawing another random number we determine whether the new edge is active or not. When the total number of elements approaches the empirical one N = 776710 we rescale the length of all edges, such that the height of the tree is T1. After this the terminal edges of the tree are extended by the time T2, such that the height of the tree is T1 + T2.
Once the phylogenetic tree is computed, we simulate the evolution of nucleotide sequences along its edges. At the root we start with the ancestral AluSx master sequence, which is mutated along the edges and duplicated at the nodes of the phylogenetic tree. Mutations are again modeled by Kinetic Monte Carlo. Nucleotides change to one of the other 3 nucleotides with rate μ0. To model the hyper-mutability of CpGs, we allow Cs and Gs in CpGs to change to T or A, respectively, with an increased rate μCpG = 6μ0.
Estimating parameters and fitting procedures
The number of Alu elements in the empirical data we estimate as the average of  over all values of k in the range 5 ≤ k ≤ 90, with L = 300. This results in N = 776710.
over all values of k in the range 5 ≤ k ≤ 90, with L = 300. This results in N = 776710.
Using the empirical data we estimate the value of the power-law exponent of the k-mer abundances distributions tail, α(k), using the Hill maximum-likelihood discrete estimator43,70 for s ≥ 3. Namely, for each k,

The obtained values of α(k) are fitted in the range 35 ≤ k ≤ 75 with Eq. (3) with  as a single free parameter [see Fig. 3] using the Levenberg-Marquardt nonlinear least squares algorithm71 in Matlab. This way we get the estimate (4).
as a single free parameter [see Fig. 3] using the Levenberg-Marquardt nonlinear least squares algorithm71 in Matlab. This way we get the estimate (4).
The value of T2 we fit manually by simulating the model with many different values of T2 with δ = 1. This results in estimate (6). With estimates (4) and (6) we simulate the model changing the value of δ. The results do not change significantly from the δ = 1 case, as long as δ is not below 5%. Furthermore, below this threshold one cannot fit the empirical estimates of α(k) with Eq. (3). Since the empirical results are well fitted with Eq. (3), we conclude that δ is the range 0.05 ≤ δ ≤ 1. The resulting set of estimated parameters used for simulations is

The results of the simulations of the model with these parameters can be seen in Fig. 2. For δ in the estimated range 0.05 ≤ δ ≤ 1 the estimates for the parameters of the model are given in Eq. (7).
Additional Information
How to cite this article: Sheinman, M. et al. Evolutionary dynamics of selfish DNA explains the abundance distribution of genomic subsequences. Sci. Rep. 6, 30851; doi: 10.1038/srep30851 (2016).
References
Ohno, S. So much “junk” DNA in our genome. In Brookhaven Symp Biol 23, 366–370 (1972).
Ponting, C. P. & Hardison, R. C. What fraction of the human genome is functional? Genome research 21, 1769–1776 (2011).
Kellis, M. et al. Defining functional DNA elements in the human genome. Proceedings of the National Academy of Sciences 111, 6131–6138 (2014).
Peng, C. et al. Long-range correlations in nucleotide sequences. Nature 356, 168 (1992).
Gao, K. & Miller, J. Algebraic distribution of segmental duplication lengths in whole-genome sequence self-alignments. PloS one 6, e18464 (2011).
Massip, F. & Arndt, P. F. Neutral evolution of duplicated DNA: an evolutionary stick-breaking process causes scale-invariant behavior. Physical review letters 110, 148101 (2013).
Massip, F., Sheinman, M., Schbath, S. & Arndt, P. F. How Evolution of Genomes Is Reflected in Exact DNA Sequence Match Statistics. Molecular biology and evolution 32, 524–535 (2015).
Mantegna, R. N. et al. Linguistic features of noncoding DNA sequences. Physical review letters 73, 3169 (1994).
Hsieh, L.-C., Luo, L., Ji, F. & Lee, H. Minimal model for genome evolution and growth. Physical review letters 90, 018101 (2003).
Sindi, S. S., Hunt, B. R. & Yorke, J. A. Duplication count distributions in DNA sequences. Physical Review E 78, 061912 (2008).
Chor, B. et al. Genomic DNA k-mer spectra: models and modalities. Genome Biol 10, R108 (2009).
Estoup, J.-B. Gammes Stenographiques (Institut Stenographique de France, 1916).
Zipf, G. K. Human behavior and the principle of least effort (Addison-Wesley Press, 1949).
Newman, M. E. Power laws, pareto distributions and Zipf’s law. Contemporary physics 46, 323–351 (2005).
Heaps, H. S. Information retrieval: Computational and theoretical aspects (Academic Press, Inc., 1978).
Gimona, M. Protein linguistics—a grammar for modular protein assembly? Nature Reviews Molecular Cell Biology 7, 68–73 (2006).
Loose, C., Jensen, K., Rigoutsos, I. & Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 443, 867–869 (2006).
Konopka, A. K., Martindale, C. et al. Noncoding dna, Zipf’s law and language. Science 268, 785–790 (1995).
Niyogi, P. & Berwick, R. C. A note on Zipf’s law, natural languages and noncoding DNA regions. arXiv preprint cmp-lg/9503012 (1995).
Chatzidimitriou-Dreismann, C., Streffer, R. & Larhammar, D. Lack of biological significance in the ‘linguistic features’ of noncoding DNA—a quantitative analysis. Nucleic acids research 24, 1676–1681 (1996).
Israeloff, N. E., Kagalenko, M. & Chan, K. Can Zipf distinguish language from noise in noncoding DNA? Physical Review Letters 76, 1976 (1996).
Bonhoeffer, S. et al. No signs of hidden language in noncoding DNA. Physical review letters 76, 1977 (1996).
Attard, G., Hurworth, A. & Jack, J. Language-like features in DNA: transposable element footprints in the genome. EPL (Europhysics Letters) 36, 391 (1996).
Tsonis, A. A., Elsner, J. B. & Tsonis, P. A. Is DNA a language? Journal of theoretical Biology 184, 25–29 (1997).
Csürös, M., Noé, L. & Kucherov, G. Reconsidering the significance of genomic word frequencies. Trends in Genetics 23, 543–546 (2007).
Doolittle, W. F. & Sapienza, C. Selfish genes, the phenotype paradigm and genome evolution. Nature 284, 601–3 (1980).
Orgel, L. E. & Crick, F. H. Selfish DNA: the ultimate parasite. Nature 284, 604–607 (1980).
Deininger, P. L. & Batzer, M. A. Mammalian retroelements. Genome research 12, 1455–1465 (2002).
Batzer, M. A. & Deininger, P. L. Alu repeats and human genomic diversity. Nature Reviews Genetics 3, 370–379 (2002).
Mighell, A., Markham, A. & Robinson, P. Alu sequences. FEBS letters 417, 1–5 (1997).
Deininger, P. L. & Batzer, M. A. Alu repeats and human disease. Molecular genetics and metabolism 67, 183–193 (1999).
Brookfield, J. F. Selection on Alu sequences? Current Biology 11, R900–R901 (2001).
Schmid, C. W. Alu: structure, origin, evolution, significance and function of one-tenth of human DNA. Progress in nucleic acid research and molecular biology 53, 283 (1996).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Matera, A. G., Hellmann, U., Hintz, M. F. & Schmid, C. W. Recently transposed Alu repeats result from multiple source genes. Nucleic acids research 18, 6019–6023 (1990).
Shen, M. R., Batzer, M. A. & Deininger, P. L. Evolution of the master Alu gene (s). Journal of Molecular Evolution 33, 311–320 (1991).
Deininger, P. L., Batzer, M. A., Hutchison, C. A. & Edgell, M. H. Master genes in mammalian repetitive DNA amplification. Trends in Genetics 8, 307–311 (1992).
Clough, J. E., Foster, J. A., Barnett, M. & Wichman, H. A. Computer simulation of transposable element evolution: random template and strict master models. Journal of molecular evolution 42, 52–58 (1996).
Johnson, L. J. & Brookfield, J. F. A test of the master gene hypothesis for interspersed repetitive DNA sequences. Molecular biology and evolution 23, 235–239 (2006).
Brookfield, J. F. & Johnson, L. J. The evolution of mobile DNAs: when will transposons create phylogenies that look as if there is a master gene? Genetics 173, 1115–1123 (2006).
Xing, J. et al. Alu element mutation spectra: molecular clocks and the effect of DNA methylation. Journal of molecular biology 344, 675–682 (2004).
Konkel, M. K. et al. Sequence analysis and characterization of active human alu subfamilies based on the 1000 genomes pilot project. Genome biology and evolution 7, 2608–2622 (2015).
Arnold, B. C. Pareto distribution (Wiley Online Library, 1985).
Cordaux, R., Hedges, D. J. & Batzer, M. A. Retrotransposition of Alu elements: how many sources? TRENDS in genetics 20, 464–467 (2004).
Prak, E. T. L. & Kazazian, H. H. Mobile elements and the human genome. Nature Reviews Genetics 1, 134–144 (2000).
Deininger, P. L., Moran, J. V., Batzer, M. A. & Kazazian, H. H. Mobile elements and mammalian genome evolution. Current opinion in genetics & development 13, 651–658 (2003).
Hedges, D. J. et al. Differential Alu mobilization and polymorphism among the human and chimpanzee lineages. Genome research 14, 1068–1075 (2004).
Kazazian, H. H. Mobile elements: drivers of genome evolution. Science 303, 1626–1632 (2004).
Slagel, V., Flemington, E., Traina-Dorge, V., Bradshaw, H. & Deininger, P. Clustering and subfamily relationships of the Alu family in the human genome. Molecular biology and evolution 4, 19–29 (1987).
Willard, C., Nguyen, H. T. & Schmid, C. W. Existence of at least three distinct Alu subfamilies. Journal of molecular evolution 26, 180–186 (1987).
Jurka, J. & Milosavljevic, A. Reconstruction and analysis of human Alu genes. Journal of molecular evolution 32, 105–121 (1991).
Kapitonov, V. & Jurkal, J. The age of Alu subfamilies. Journal of molecular evolution 42, 59–65 (1996).
Price, A. L., Eskin, E. & Pevzner, P. A. Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome research 14, 2245–2252 (2004).
Churakov, G. et al. A novel web-based tint application and the chronology of the primate Alu retroposon activity. BMC evolutionary biology 10, 376 (2010).
Willis, J. C. & Yule, G. U. Some statistics of evolution and geographical distribution in plants and animals and their significance. Nature 109, 177–179 (1922).
Sheinman, M., Massip, F. & Arndt, P. F. Statistical Properties of Pairwise Distances between Leaves on a Random Yule Tree. PLoS One 10 (2015).
Zuckerkandl, E. & Pauling, L. Molecular disease, evolution and genetic heterogeneity 189–225 (Academic Press, 1962).
Kumar, S. Molecular clocks: four decades of evolution. Nature Reviews Genetics 6, 654–662 (2005).
Li, W.-H., Tanimura, M. & Sharp, P. M. An evaluation of the molecular clock hypothesis using mammalian DNA sequences. Journal of molecular evolution 25, 330–342 (1987).
Scally, A. & Durbin, R. Revising the human mutation rate: implications for understanding human evolution. Nature Reviews Genetics 13, 745–753 (2012).
Labuda, D. & Striker, G. Sequence conservation in Alu evolution. Nucleic acids research 17, 2477–2491 (1989).
Batzer, M. A. et al. Structure and variability of recently inserted Alu family members. Nucleic acids research 18, 6793–6798 (1990).
Liu, G. E., Alkan, C., Jiang, L., Zhao, S. & Eichler, E. E. Comparative analysis of Alu repeats in primate genomes. Genome research 19, 876–885 (2009).
Hedges, S. B., Marin, J., Suleski, M., Paymer, M. & Kumar, S. Tree of life reveals clock-like speciation and diversification. Molecular Biology and Evolution 32, 835–845 (2015).
Britten, R. J. Evidence that most human Alu sequences were inserted in a process that ceased about 30 million years ago. Proceedings of the National Academy of Sciences 91, 6148–6150 (1994).
Cunningham, F. et al. Ensembl 2015. Nucleic acids research 43, D662–D669 (2015).
Karolchik, D. et al. The ucsc table browser data retrieval tool. Nucleic acids research 32, D493–D496 (2004).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Bortz, A. B., Kalos, M. H. & Lebowitz, J. L. A new algorithm for Monte Carlo simulation of Ising spin systems. Journal of Computational Physics 17, 10–18 (1975).
Clauset, A., Shalizi, C. R. & Newman, M. E. Power-law distributions in empirical data. SIAM review 51, 661–703 (2009).
George, A., Seber, F. & Wild, C. Nonlinear regression (Wiley-Interscience, 2003).
Austen, J. Pride and prejudice (Lulu. com, 1996).
Acknowledgements
We thank M.A. Batzer, A. Mammana, A. Fratzl and R. Hermsen for fruitful discussions and help.
Author information
Authors and Affiliations
Contributions
M.S. and P.F.A. analysed the data. All authors designed the research and wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sheinman, M., Ramisch, A., Massip, F. et al. Evolutionary dynamics of selfish DNA explains the abundance distribution of genomic subsequences. Sci Rep 6, 30851 (2016). https://doi.org/10.1038/srep30851
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep30851
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
