
Abstract
There is growing evidence that microRNAs are implicated in pulmonary arterial hypertension (PAH), but underlying mechanisms remain elusive. Here, we identified that miR-223 was significantly downregulated in chronically hypoxic mouse and rat lungs, as well as in pulmonary artery and pulmonary artery smooth muscle cells (PASMC) exposed to hypoxia. Knockdown of miR-223 increased PASMC proliferation. In contrast, miR-223 overexpression abrogated cell proliferation, migration and stress fiber formation. Administering miR-223 agomir in vivo antagonized hypoxia-induced increase in pulmonary artery pressure and distal arteriole muscularization. RhoB, which was increased by hypoxia, was identified as one of the targets of miR-223. Overexpressed miR-223 suppressed RhoB and inhibited the consequent phosphorylation of myosin phosphatase target subunit (MYPT1) and the expression of myosin light chain of myosin II (MLC2), which was identified as another target of miR-223. Furthermore, serum miR-223 levels were decreased in female patients with PAH associated with congenital heart disease. Our study provides the first evidence that miR-223 can regulate PASMC proliferation, migration and actomyosin reorganization through its novel targets, RhoB and MLC2, resulting in vascular remodeling and the development of PAH. It also highlights miR-223 as a potential circulating biomarker and a small molecule drug for diagnosis and treatment of PAH.
Similar content being viewed by others


Introduction
Chronic hypoxia-induced pulmonary arterial hypertension (PAH) is characterized by persistent elevation of pulmonary arterial pressure due to pulmonary vasoconstriction and pulmonary vascular remodeling accompanied by right ventricular hypertrophy1. It is well accepted that the primary cellular mechanism underlying vascular remodeling is the excessive proliferation and migration of pulmonary arterial smooth muscle cells (PASMC)2,3. However, the exact molecular mechanisms that regulate vascular proliferation are incompletely understood.
Currently, signaling pathways, such as the Rho-kinase pathway, are implicated as important factors in the pathogenesis of PAH1. RhoA has been widely reported as a regulator of actomyosin-related processes such as cell adhesion, migration and proliferation4. Recent data show that RhoB is highly expressed in lung tissue and is involved in cellular responses to oxidative stress5,6. Decrease in RhoB markedly inhibits hypoxia-induced endothelial permeability and growth responses in pulmonary vascular cells. Moreover, genetic deletion of RhoB attenuates development of chronic hypoxia-induced vascular remodeling and pulmonary hypertension in mice5.
MicroRNAs (miRNAs) have been identified as essential modulators of a variety of genes and cellular processes7. Several miRNAs have been reported to regulate different steps in the process of angiogenesis and vascular remodeling8. miR-21 negatively regulates programmed cell death 4 (PDCD4) promoting PASMC proliferation and migration9,10. Downregulation of miR-204 correlates with PAH severity and accounts for the pro-proliferative and anti-apoptotic phenotypes of PASMC in PAH11. miR-143 and miR-145 have been demonstrated to play critical roles in vascular smooth muscle cell phenotype switching12,13. Decreased expression of miR-124 affects the proliferation, migration and inflammatory phenotype of both PASMC and pulmonary vascular fibroblasts14,15. Alterations of the miR-210–iron–sulfur cluster assembly homolog 1/2 (ISCU1/2) regulatory axis can cause Fe-S deficiencies in vivo and is crucial to the development of PAH16. miR-17~92 inhibits PDZ and LIM domain 5 (PDLIM5) to induce the transforming growth factor-beta 3/ SMAD family member 3 (TGF-beta3/SMAD3) pathway, contributing to the pathogenesis of PAH17. Treatment with antagomiR-20a restores the functional levels of bone morphogenetic protein receptor II (BMPR2) in pulmonary arteries and prevents the development of vascular remodeling18. miRNAs are now recognized as participants in multiple disease pathways in the pulmonary vasculature, however their true importance in PAH is just beginning to emerge19.
miR-223 was initially described as a myeloid-specific miRNA20,21 and as a biological marker for several tumoral processes22,23,24. It has also been reported to play an important role in neuroprotection25 and as a marker of damage to both skeletal26 and cardiac muscle tissues20. Recent studies have indicated that miR-223 is an anti-angiogenic miRNA that prevents endothelial cell proliferation, migration and sprouting by targeting β1 integrin27. In this study, we elucidated the role of miR-223 in pulmonary vascular responses to hypoxia in vitro and in vivo and identified a RhoB/ myosin light chain-2 (MCL2)-dependent role of miR-223 in PASMC and its clinical importance as a potential biomarker for the diagnosis of pulmonary arterial hypertension associated with congenital heart disease (CHD-PAH).
Results
Hypoxia decreases miR-223 expression in lungs, pulmonary artery and pulmonary arterial smooth muscle cells
To identify miRNAs that are altered during the development of chronic hypoxia-induced pulmonary hypertension, we compared miRNA expression levels in normoxic and chronically hypoxic mouse lungs by microarray analysis. The results revealed that standing out from 1040 miRNAs measured, there were five significantly changed miRNAs between normoxia and hypoxia (p < 0.01) and miR-223 (also called miR-223-3p) was the most significantly downregulated miRNA (Fig. 1A). An independent quantitative real-time PCR (qRT-PCR) assay confirmed the decreased expression of miR-223 in mouse lungs in response to hypoxia (Fig. 1B). The decrease in miR-223 was also observed in rat lungs and in pulmonary arteries (PA) following 3-weeks of hypoxia (Fig. 1C,D, respectively).

miR-223 is downregulated by hypoxia in PASMC.
(A) Differential expression of candidate miRNAs in mouse lung tissue exposed to normoxia (21% O2)(Con) or hypoxia (10% O2) for 2 days (H-2d), 1 week (H-1w), 2 weeks (H-2w) and 3 weeks (H-3w) (n = 3 or 4 for each group). Cluster analysis of miRNA expression in a 1040 miRNA microarray set measured from individual specimens (p < 0.05 compared with normoxic controls). Validation of miR-223 downregulation by hypoxia in mouse lungs (B), rat lungs, n = 8 (C) and rat pulmonary arteries (PA), n = 8 (D) was performed by qRT-PCR. (E) Rat and human PASMC were exposed to normoxia (21% O2) (N-Con) or hypoxia (H, 3% O2) (n = 3) for up to 48 h. The relative levels of miR-223 were measured by real-time PCR using SNORD44 (for human sample) or snoRNA202 (for mouse or rat sample) as an internal control. Experiments were performed in triplicate. Bar charts show the relative expression level by normalizing to the normoxia control (N-Con). Data are shown as means ± SE, *p < 0.05, **p < 0.01.
To determine whether miR-223 expression in PASMC parallels expression in lungs of hypoxic mice, we isolated PASMC from Sprague-Dawley rats and exposed them to hypoxia. We observed a 50% decrease in miR-223 levels, with a maximal decrease after 6 to 12 hours of hypoxia (Fig. 1E, left panel). Comparable results were obtained when expression of miR-223 was studied in human PASMC (hPASMC) under hypoxic conditions (Fig. 1E, right panel). These results point to miR-223 as a miRNA that is decreased by hypoxia in PASMC.
MiR-223 represses pulmonary smooth muscle cell proliferation and migration
Pulmonary vascular remodeling in response to chronic hypoxia is principally associated with the excessive proliferation and migration of PASMC2,28. To assess the role of miR-223 in modulating cell proliferation, cells were transfected with miR-223 mimic, which increased miR-223 levels in cultured rPASMC and hPASMC (Supplementary Figure S1). EdU incorporation assay and PCNA immunoblotting showed that miR-223 overexpression significantly prevented the increase in cell proliferation induced by hypoxia (Fig. 2A,B). To confirm the consequences of miR-223 on cell proliferation, EdU incorporation assay and PCNA immunoblotting were also performed in cells transfected with miR-223 inhibitor. Conversely, miR-223 inhibition augmented the proliferation of PASMC under normoxic conditions (Supplementary Figure S2A, B).

miR-223 inhibits PASMC proliferation, migration and stress fiber formation.
Rat and human PASMC were transfected with mimic control (miR-Con) or miR-223 mimic (miR-223) and exposed to hypoxia (3% O2) or normoxia for 24 h in triplicate. Cell proliferation was determined by EdU incorporation assay (A) and PCNA immunoblotting (B). Data are shown as means ± SE (n = 3). (C) Transwell migration assay showing the migration of rPASMC or hPASMC with miR-223 overexpression. Bar charts representing relative migrated cells after 24 h. Data (mean ± SE) is derived from an average of three independent experiments, with five visual fields in triplicate samples. (D) F-actin in cells was stained with TRITC-phalloidin and visualized by confocal microscopy. *p < 0.05, **p < 0.01.
Furthermore, enhanced expression of miR-223 also inhibited hypoxia-induced increase in cell migration in both rPASMC and hPASMC, as measured using a chamber invasion assay of cells passing through a transwell filter (Fig. 2C). Moreover, the altered migration in miR-223-expressing cells was associated with a decrease in stress fiber levels, a feature reflected by the higher F-actin content in cells that have active actomyosin contraction (Fig. 2D). All the results above indicate that miR-223 is involved in the proliferative and migratory responses in PASMC to hypoxia.
MiR-223 attenuates chronic hypoxia-induced pulmonary vascular remodeling
To confirm our findings in vivo, we determined the effect of administering an agomir of miR-223 or an unrelated agomir control on right ventricular systolic pressure (RVSP) and pulmonary vascular muscularization in rats exposed to chronic hypoxia. Following 3 weeks of hypoxia exposure, the RVSP in wild-type rats increased from 12.33 ± 1.75 mmHg to 20.14 ± 2.58 mmHg (p < 0.01). Rats with miR-223 agomir treatment showed a significantly attenuated increase in RVSP in hypoxia (15.81 ± 3.00 mmHg in miR-223 compared with 21.06 ± 2.54 in miR-Con, p < 0.05) (Fig. 3A). Coincident with the effects on pulmonary vascular pressure, the ratio of right to left ventricle plus septum weight [RV/(LV + S)] in rats treated with miR-223 agomir also showed an attenuated increase in hypoxia (Fig. 3B). RVSP or RV/(LV + S) in rats treated with agomir control were not different compared with hypoxic control group. Next, we measured miR-223 levels in peripheral blood from each experimental group by qPCR. Serum miR-223 levels were significantly decreased in hypoxia-exposed rats (p < 0.05, compared with normoxia group) and the levels were increased with miR-223 agomir administration (p < 0.05, compared with miR-Con group in hypoxia) (Fig. 3C). As pulmonary vascular remodeling is a characteristic feature of PAH19,29, we noted that the delivery of synthetic miR-223 agomir markedly reduced hypoxia-increased medial wall thickness of small and medium sized pulmonary arteries (Fig. 3D,E). Our results show that pulmonary artery pressure and distal arteriolar muscularization following chronic hypoxia were significantly decreased when level of miR-223 was preserved.

miR-223 attenuates chronic hypoxia-induced pulmonary vascular remodeling.
Chronic hypoxia-induced PAH in a rat model was established by 3 weeks of hypoxic treatment. Rats were injected with either agomir control (miR-Con) or agomir specifically expressing miR-223 (miR-223). Right ventricular systolic pressure (RVSP) (A) and right ventricular hypertrophy [RV/ (LV + S)] (B) are shown in bar charts. Data are shown as means ± SE (n = 8). (C) The relative levels of serum miR-223 in each rat group were measured by real-time PCR using rnomiR-16-5p as a normalization control. Data are shown as means ± SE (n = 8). (D,E) The wall thickness of small and medium sized pulmonary arteries was assessed from HE stained lung sections. Scale bar = 50 μm. Bar charts showing the percentage of wall thickness to vessel external diameter (WT%) of each rat group. Data are shown as means ± SE (n = 8), *p < 0.05, **p < 0.01.
MiR-223 targets RhoB and MLC2 in pulmonary arterial smooth muscle cells
To determine how miR-223 regulates PASMC proliferation, migration and consequently pulmonary vascular remodeling, we identified the miR-223 target proteins involved in these processes. A negative correlation between the expression of miR-223 and RhoB protein was detected in mouse lungs, as well as in rat and human PASMC exposed to hypoxia (Fig. 4A). The mRNA level of RhoB in both hypoxia-treated rPASMC and hPASMC was also negatively correlated with miR-223 expression (Fig. 4B).

Hypoxia upregulates RhoB/Rho-kinase signaling in PASMC.
(A) Representative western blot for hypoxia-induced increase of RhoB in mouse lungs, rat PASMC and human PASMC (n = 3). β-actin was used as a loading control. (B) RhoB mRNA levels in hypoxia-exposed rat and human PASMC were evaluated by qRT-PCR using β-actin as an internal control. Bar charts showing the relative expression level by normalizing to the normoxia control (N-Con). Data are shown as means ± SE (n = 3). (C) Expression of RhoB/Rho-kinase signaling pathway related components in rat and human PASMC in response to hypoxia was determined by western blotting (n = 3). *p < 0.05, **p < 0.01.
Rho-kinase (ROCK) is a direct downstream effector of Rho GTPase (such as RhoA and RhoB). It phosphorylates the regulatory subunit of myosin phosphatase, known as MYPT1 and inhibits its phosphatase activity. This inhibition increases phosphorylation of MLC, which is thought to induce Rho-mediated assembly of stress fibers and focal adhesions30. Hypoxia is a common stimulus for activating Rho/ROCK and thereby facilitates the phosphorylation of MYPT1 and MLC30,31. Here, we observed increased levels of phosphorylated MYPT1 and MLC2 in both rPASMC and hPASMC following exposure to hypoxia (Fig. 4C).
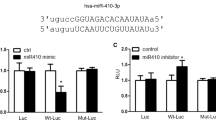
Using an online tool (TargetScan, www.targetscan.org), we found two potential miR-223 binding sites within RhoB 3′-UTR (Fig. 5A). In order to validate RhoB as a direct target of miR-223, we performed a reporter gene assay to ascertain the interactions between miR-223 and RhoB 3′-UTR. Co-transfection of HEK-293 cells with pre-miR-223 and a luciferase construct containing the two putative miR-223 binding sequences within the 3′-UTR of RhoB resulted in a significant decrease (about 50%, p < 0.01) in luciferase activity, indicating direct interaction between them. Notably, the two target sites when tested in the assay exhibited substantial differences in their susceptibility to miR-223 mediated repression. Mutation of the downstream binding site (Mut 2) completely antagonized the inhibitory effect of miR-223 in the luciferase assay. However, effect was unchanged when the target site 1 sequence was mutated (Fig. 5A). The results above clearly show that RhoB is a direct target of miR-223 in PASMC. Rho GTPase activation assay showed that miR-223 not only repressed the expression of RhoB but also markedly reduced its activity (Fig. 5C). miR-223 inhibition also enhanced the phosphorylation of MYPT1 and MLC2, partially mimicking the effects of hypoxia. Conversely, overexpression of miR-223 reduced the levels of phosphorylated MYPT1 and MLC2 (Fig. 5D). Notably, a negative correlation of total MLC2 expression and miR-223 levels was found in rPASMC and hPASMC (Fig. 5D). miRNA target prediction with FINDTAR3 indicated a potential miR-223 binding site in the 3′-UTR of MLC2. Reporter gene assay revealed a direct interaction between miR-223 and the 3′-UTR of MLC2, which was confirmed by binding site mutants in reporter assay (Fig. 5B). Results above demonstrate that miR-223 contributes to hypoxia-induced phosphorylation of MYPT1 by targeting RhoB and more importantly, has a direct inhibitory effect on MLC2 expression. In addition, we measured the protein levels of these two targets in rats with hypoxia-induced PAH with miR-Con and miR-223 agomir treatment. As shown in Fig. 5E, both RhoB and MLC2 proteins in hypoxia-treated rat lungs were increased, but to a significantly lesser degree in rats treated with synthetic miR-223 agomir when compared to agomir control. We observed similar results with the cell proliferative marker PCNA, indicating that miR-223 has an anti-proliferative effect in experimental PAH.

Identification of RhoB and MLC2 as direct targets of miR-223.
Luciferase reporter gene assay on the interaction between miR-223 and 3′-UTR of RhoB (A) and MLC2 (B) in HEK293 cells. Upper panel: putative miR-223 binding sites in the 3′-UTR of RhoB or MLC2 along with the mutation sites. TS1,target site 1 (a pooly conserved binding site); TS2, target site 2 (a conserved binding site); Bottom panel: 3′-UTR luciferase reporter assay with targets and their mutant along with miR-223/miR-Con overexpressing vectors. Bar charts of luciferase reporter analysis represent means ± SE (n = 3), firefly luciferase activity was normalized to renilla luciferase activity. (C) Active RhoB proteins in miR-223 or miRNA control overexpressing PASMC were pulled down by rhotekin-RBD beads and detected by western blotting analysis using anti-RhoB antibody (n = 3). (D) Expression of RhoB/Rho-kinase signaling pathway related components in PASMC, with overexpression or inhibition of miR-223, was measured by western blotting (n = 3). (E) Expression of RhoB, MLC2 and PCNA in lungs of hypoxia-exposed rats administered agomirs miR-223/miR-Con was determined by western blotting (n = 3). β-actin was used as a loading control for the above western blot analysis. Two representative blots are shown. *p < 0.05, **p < 0.01.
Serum miR-223 is a potential circulating biomarker for PAH
As serum miRNAs are currently used as potential biomarkers for human diseases32,33,34, we wondered if circulating miR-223 levels were decreased in patients with PAH akin to that in experimental PAH and human PASMC. First, we measured miR-223 levels in sera from 30 patients with CHD-PAH, mainly with ventricular septal defect (VSD) or atrial septal defects (ASD) (Supplementary Table 1). We found that serum miR-223 levels in patients were lower than that in healthy donors but with no significant difference (p = 0.058) (Fig. 6A). As miR-223 locus is located on the X chromosome, a new study was carried out on 13 male CHD-PAH patients/ healthy donors or 17 female CHD-PAH patients/ healthy donors to determine whether gender differences exist (Supplementary Table S1). Our results show that the levels of circulating miR-223 in CHD-PAH manifest a gender related difference. Serum levels of miR-223 in female CHD-PAH patients were significant lower than that in healthy female donors, but there was no difference between male CHD-PAH subjects and control subjects (Fig. 6B). Another study on 7 male and 17 female CHD-PAH patients/ healthy donors (Supplementary Table S2) showed significant decrease in circulating miR-223 levels in CHD-PAH patients and confirmed the gender-related alteration (Fig. 6C,D). These results are of great clinical significance as it highlights miR-223 as a potential diagnostic circulating biomarker in female CHD-PAH patients.

Serum miR-223 as a potential circulating biomarker for CHD-PAH.
Expression of serum miR-223 was measured by qRT-PCR in healthy human donors (normal) and CHD-PAH patients. Levels of miR-223 in 30 normal versus 30 CHD-PAH patients (A), 13 male normal versus 13 male CHD-PAH patients and 17 female normal versus 17 female CHD-PAH patients (B), a second group of 24 normal versus 24 patients (C), 7 male normal versus 7 male patients and 17 female donors versus 17 female patients (D), respectively were normalized to hsa-miR-16-5p and represented in scatter plots. Data are shown as means ± SE, **p < 0.01.
Discussion
This study reveals that miR-223 is rapidly downregulated in response to hypoxia in PASMC and is critical to chronic hypoxia-associated pulmonary vascular pathology, such as smooth muscle cell proliferation, migration and actomyosin reorganization, which ultimately results in vascular remodeling and distal arteriole muscularization. We elucidate a RhoB/ROCK-associated regulatory pathway that is suppressed by miR-223 and the decrease of miR-223 in hypoxia leads to pathologic changes in PASMC (as summarized in Fig. 7). This study identifies miR-223 as a potential target in experimental PAH and the possible benefits of miR-223 agomir therapy in its treatment. It also suggests a potential use of miR-223 levels as a circulating biomarker for clinical diagnosis in women with CDH-PAH.

Schematic model showing the role of miR-223 in pulmonary arterial hypertension in response to hypoxia.
miR-223 attenuates hypoxia-induced smooth muscle cell proliferation, migration and actomyosin reorganization and the consequent vascular remodeling and pulmonary hypertension by regulating the RhoB/MLC pathway.
Hypoxic pulmonary vasoconstriction helps to maintain oxygen supply within physiological limits by diverting blood to the poorly ventilated areas of the lung35. A specific and still growing list of miRNAs, so-called “hypoxamirs”, are characterized to be dynamically regulated by hypoxia36. In this study, we discovered a decrease in expression of miR-223 in hypoxia-induced PAH mouse lungs, pulmonary artery and isolated PASMC. In an early study, Caruso et al. reported that miR-223 was decreased in chronic hypoxia-treated PAH rat lungs37. Recently, Shi et al. reported that miR-223 antagonizes angiogenesis by abrogating VEGF and bFGF-induced cell proliferation, migration and tube formation in vascular endothelial cells27. Rangrez et al. found that overexpressing miR-223 in aortic vascular smooth muscle cells (VSMCs) increased proliferation and markedly enhanced cell migration38, demonstrating miR-223 may function in a tissue-specific manner. Our data show that increasing the expression of miR-223 abrogated the hypoxia-induced increase in proliferation, migration, as well as stress fiber assembly in both rPASMC and hPASMC. Overexpression of miR-223 using agomir antagonized the hypoxic effects on pulmonary artery pressure and distal arteriolar muscularization in vivo. DNA-damage induced PARP-1 has been shown to be involved in PAH-PASMCs and interestingly baseline DNA-damage was demonstrated to be higher in PAH patients and their relatives39. During the preparation of this manuscript, Meloche et al. most recently showed that miR-223 was downregulated in PAH-PASMCs and lungs of monocrotaline-treated PAH rats. They reported that miR-223 played anti-proliferative and pro-apoptosis roles in PAH-PASMC by directly repressing PARP-140. Our study demonstrates that miR-223 regulation directly targets RhoB and MLC2 to affect vascular remodeling and hypoxia-induced pulmonary hypertension.
Both RhoA and RhoB appear to cooperate in hypoxia-induced cytoskeletal remodeling, but RhoB plays a unique role in the regulation of vascular proliferation responses to hypoxia5. Recent reports implicate RhoB as a regulator of hypoxia-induced cell proliferation in both human pulmonary artery endothelial cells and hPASMC. Moreover, RhoB deficiency in mice markedly attenuated development of chronic hypoxic pulmonary hypertension, despite compensatory expression of RhoA in the lung5. Stothard et al. have reported that RhoB protein levels and activity in hPASMC rapidly increased to maximum between 2 to 4 hours in 2% O2, while RhoA mRNA and protein levels remained relatively unchanged with a small increase in its activity. However in this study, we found that RhoB mRNA levels in hPASMC, when exposed to a milder hypoxic condition (3% O2), reached a maximum between 6 to 12 hours, with protein levels continuing to rise until 24 hours. In cultured rPASMC, RhoB gene and protein expression increased steadily after hypoxic exposure and reached maximum levels at 6 h and 24 h respectively. These hypoxia-induced changes in RhoB were opposite to the effects on miR-223, pointing to a negative relationship between them.
The correlation between the expression of RhoB and miR-223 was ascertained in primary culture of PASMC and in an in vivo PAH rat model by both miR-223 overexpression and/or knockdown approaches. It was previously reported that the 3′-UTR of RhoB could be targeted by hsa-miR-223 in human cell lines41. Here we further corroborated RhoB as a direct target of rno-miR-223 in rat PASMC. TargetScan predicted RhoB having two miR-223 target sites and defined site 1 as poorly conserved and site 2 as a conserved site. Reporter gene analysis revealed that mutation of site 2 resulted in complete loss of the inhibitory activity of miR-223, while site 1 mutation showed no activity change compared to wild type construct, although it indicates that there may be more bases pairing with miR-223. The results fit well with a recent report that hsa-miR-223 interacts with RhoB, in which the authors proposed that some AU-rich motif located upstream of distal miR-223-binding site enhances the miRNA function, independent of the miRNA target sequence being tested41.
Previous studies have demonstrated that constitutively activated Rho proteins protect against the disruption of stress fibers and control actomyosin contractility in both smooth muscle and non-muscle cells42. MLC phosphorylation is a critical step that leads to activation of myosin ATPase and subsequent smooth muscle cell contraction in response to hypoxia. MLC is phosphorylated by MLC kinase (MLCK) and de-phosphorylated by MLC phosphatase (MLCP). ROCK, a specific downstream effector of Rho GTPase, increases the levels of phosphorylated MLC by direct phosphorylation and also through the inhibition of MLCP by phosphorylating its regulatory subunit (MYPT1)30,43. Our data confirm that hypoxia resulted in a significant increase in phosphorylation of MYPT1 and MLC. Total MYPT1 and MLC2 protein levels were not different when compared with that in normoxic conditions. Overexpression of miR-223 caused a remarkable decrease in MLC2 protein levels, while miR-223 inhibition increased the expression of MLC2. Further, we demonstrated the 3′-UTR of MLC2 mRNA was post-transcriptionally regulated by miR-223. Our study has uncovered for the first time a regulatory miRNA that directly targets RhoB and MLC2 in the Rho/ROCK signaling pathway (Fig. 7).
Due to emerging evidence for a role of Rho proteins in pulmonary hypertension, specific and potent modulators of various steps of Rho GTPase signaling pathway are becoming promising means for pharmacological intervention. ROCK inhibitors, such as Fasudil and Y-27632, are examples of two widely used chemical compounds that have been developed to treat hypoxia-induced pulmonary vascular remodeling. The success of the ROCK inhibitors in the treatment of vascular disease has encouraged the use of Rho pathway inhibition in clinical studies. On the other hand, there are few effective inhibitors directly targeting Rho GTPases, likely owing to the lack of optimal structural information on interaction with individual GTPase to achieve specificity. However, the dynamic changes of Rho GTPase in disease states underline the need to develop effective inhibitors for therapeutic applications. Here, we discovered that treatment with miR-223 agomir markedly attenuates chronic hypoxia-induced pulmonary vascular remodeling in vivo, due to its post-transcriptionally inhibitory effects on RhoB and MLC2 expression. Therefore, a small molecular drug, like miR-223, may improve efficacy by targeting the RhoB/Rho Kinase/MLC2 signaling in pulmonary vascular diseases.
There is increasing evidence that circulating miRNAs may be informative in disease diagnosis and have a potential to be used as biomarkers of disease44,45. Stable miRNAs in the circulation are speculated to exist in microvesicles or exosomes and apoptotic bodies, which are secreted into the circulation and are extremely stable in the plasma or serum46,47. miR-223 is currently of interest as its levels in circulation are inversely correlated with the risk of developing coronary heart disease, diabetes and myocardial infarction48,49,50. A recent study on circulating miRNAs as potential markers for pulmonary hypertension by using a microarray approach identified that plasma miR-223 levels were decreased in these patients34. In our study, we uncovered that there are gender differences in downregulation of serum levels of circulatory miR-223 in patients with CHD-PAH. However, no gender difference was observed in healthy donors. Torres et al. reported that patients with chronic obstructive pulmonary disease (COPD) also manifested gender related differences in some plasma inflammatory cytokines51. Circulating miRNAs are speculated to be released from the damaged lungs or vessels in patients with pulmonary hypertension34. Recently, Ma reported that miR-27b was greatly up-regulated in the lung of CHD-PAH patients and correlated well with preoperative mean pulmonary arterial pressure52. Our results point to a gender-specific response in circulating miR-223 in pulmonary hypertension associated with CHD. It still remains unknown if there is a relationship between miR-223 reduction in female serum and gene regulatory abnormality on X chromosome in CHD-PAH patients.
The data presented are based on a limited number of subjects and the results need to be further validated in a larger cohort of the disease. We have not explored the miR-223 expression and RhoB/ROCK/MLC pathway in pulmonary artery endothelial cells (PAECs), the abnormality of which also contributes to the pathology of pulmonary arterial hypertention. It will be of great interest to study whether miR-223 agomir shows better effect by intratracheal nebulization.
Materials and Methods
Cell culture
Human HEK293 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Human pulmonary arterial smooth muscle cells (hPASMC) were purchased from Lonza (Walkersville, MD) and cultured in SmGM-2 smooth muscle growth media consisting with smooth muscle basal medium, 5% FBS, 0.5 ng/ml human recombinant epidermal growth factor, 2 ng/ml human recombinant fibroblast growth factor, 5 μg/ml insulin and 50 μg/ml gentamicin.
Rat pulmonary arterial smooth muscle cells (rPASMC) were dissociated from the pulmonary artery of Sprague-Dawley rats (male, 6 weeks). Briefly, a segment of pulmonary artery just proximal to the lung entry was removed under aseptic conditions and cleaned from connective and fat tissues; The media of the pulmonary artery, dissected from the adventitia and intima, was subjected to anenzymatic digestion in buffer containing 1 mg/ml collagenase (Worthington Biochemical Corporation) and 0.5 mg/ml Elastase type IV (Sigma-Aldrich) for 60 min at 37 °C. For hypoxic treatment, PASMC were placed in a special hypoxia incubator infused with a gas mixture of 5% CO2 and nitrogen to obtain 3% oxygen concentration. Oxygen concentration was monitored continuously (Forma 3130; Thermo Scientific).
miRNA microarray assay
Microarray assay of mature miRNAs was performed by the LC Sciences Microarray Service (LC Sciences, Houston, TX). Briefly, total RNA (5 μg) was size fractionated (<300 nucleotides) by using a YM-100 Microcon centrifugal filter (Millipore) and the small RNAs (<300 nt) isolated were polyadenylated with poly(A) polymerase. An oligonucleotide tag was then ligated to the poly(A) tail for later fluorescent dye staining. Each sample was hybridized to a microarray that contained 1040 mature mouse miRNA probes from the Sanger miRBase 16.0 (http://www.sanger.ac.uk/Software/Rfam/mirna/). After RNA hybridization, tag-conjugating Cy3 and Cy5 dyes were circulated through the microfluidic chip for dye staining. Fluorescence images were collected using a laser scanner (GenePix 4000B, Molecular Device) and digitized using Array-Pro image analysis software (Media Cybernetics).
Data were analyzed by first subtracting the background and then normalizing the signals using a LOWESS filter (Locally-Weighted Regression) for two color experiments. Transcripts were determined as detectable if their signal intensity was higher than 3 times the background standard deviation, spot CV (standard deviation/signal intensity) was <0.5 and transcripts had at least 50% of repeating probe signals above the detection level. P-values of the ANOVA were calculated. The miRNAs significantly expressed in hypoxia-treated mice compared with normoxic control (p values <0.05) were selected for cluster analysis according to a hierarchical method, which was performed with average linkage and Euclidean distance metrics. The clustering plot was generated using TIGR MeV (Multiple Experimental Viewer) software from the Institute for Genomic Research.
miR-223 overexpression and inhibition
Chemically synthesized miRNA mimics or inhibitors to overexpress or inhibit miR-223 and each unrelated negative control (miR-Con or anti-Con), were purchased from Ribobio (Guangzhou, China). Cells at 70% confluence after overnight culture on petri dishes were transfected with miR-223 mimic, mimic control (miR-Con) (50 nmol/L), or miR-223 inhibitors (anti-223), inhibitor control (anti-Con) (100 nmol/L) by Lipofectamine 2000 (Invitrogen). After 6 h transfection, medium was changed and the cells were cultured for 24 h and then exposed to either hypoxic or normoxic conditions for 24 h or 48 h.
Quantitative RT-PCR
Total RNA of tissue and serum sample was extracted with RNAiso Plus (TaKaRa Biotechnology, China). For mRNA evaluation, 1 μg of total RNA was reverse-transcribed using M-MLV Reverse Transcriptase (TaKaRa) with oligo(dT)18 plus random hexamer primers (Promega). qRT-PCR was performed with gene specific primers and SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) on ABI StepOneplus real-time PCR System (Applied Biosystems). The expression level of each gene was normalized to internal control β-actin gene and the expression level of each mRNA was calculated using the 2(−ΔΔCt) method. For miRNA assay, the mature miR-223 expression level was determined according to S-Poly(T) method, as we previously reported53,54. The miR-223 expression level was normalized to SNORD44 (in human tissue sample), rno-miR-16-5p (in rat serum sample) or snoRNA202 (in mice or rat tissue sample) and calculated using the 2(−ΔΔCt) method. The miR-223 expression level in human serum samples was normalized to hsa-miR-16-5p and calculated using the 2(−ΔCt) method. Each PCR reaction was analyzed in triplex tubes. Primers used for reverse transcriptionand quantitative PCR were listed as follows: human RhoB: 5′-CGG ACT CGC TGG AGA ACA-3′ (forward) and 5′-GAG GTA GTC GTA GGC TTG GAT-3′ (reverse); rat RhoB: 5′-CGG ACT CTC TCG AGA ACA-3′ (forward) and 5′-GAG GTA GTC ATA GGC TTG GAT-3′ (reverse); rat β-actin: 5′-AAA GAC CTG TAC GCC AAC AC-3′ (forward) and 5′-GTC ATA CTC CTG CTT GCT GAT-3′ (reverse); human β-actin: 5′-AGA GAT GGC CAC GGC TGC TT-3′ (forward) and 5′-ATT TGC GGT GGA CGA TGG AG-3′ (reverse); miR-223: 5′-GTG CAG GGT CCG AGG TCA GAG CCA CCT GGG CAA TTT TTT TTT TTG G-3′ (RT) and 5′-GGT GTC AGT TTG TCA AAT ACC C-3′ (forward); miR-16: 5′-GTG CAG GGT CCG AGG TCA GAG CCA CCT GGG CAA TTT TTT TTT TTC GCC AA-3′ (RT) and 5′- CCG GGT AGC AGC ACG TAA ATA-3′ (forward); SNORD44: 5′-GTG CAG GGT CCG AGG TCA GAG CCA CCT GGG CAA TTT TTT TTT TTA GTC AG-3′ (RT) and 5′-TGG CCT GGA TGA TGA TAA GCA-3′ (forward); snoRNA202: 5′-GTG CAG GGT CCG AGG TCA GAG CCA CCT GGG CAA TTT TTT TTT TTC ATC AG-3′ (RT) and 5′- GTA CTT TTG AAC CCT TTT CCA T-3′ (forward).
Cell proliferation assay
Cell proliferation was determined by EdU incorporation assay and PCNA immunoblotting. EdU labeling was performed by using the EdU Assay Kit (Ribobio, Guangzhou, China) following the procedures suggested by the manufacturer. Briefly, approximately 1 × 104 cells were seeded in triplicates in 48-well plates and the cells were cultured for 24 h and then exposed to either hypoxic or normoxic conditions for 24 h, then exposed to 20 μmol/L EdU for 24 h at 37 °C. The cells were then fixed in 4% paraformaldehyde for 30 min at room temperature and permeabilized in 0.5% Triton X-100 for 10 min. Cells were washed with PBS and each well was incubated with 200 μl 1× Apollo reaction cocktail for 30 min. DNA was then stained with 1 μg/ml DAPI (200 μl per well) for 5 min and imaged under a fluorescent microscope. All data are shown as a percentage of normoxia controls. PCNA Western blot studies were performed as described in Western blotting. All experiments were performed in triplicate.
Transwell Cell migration assay
Cells were seeded on the upper chambers of a 24 well format cell culture insert with 8 μm pores (FALCON, Le Pont De Claix, France). Upper Medium was replaced by starvation medium (containing 0.5% FBS), while the lower chamber contained medium with 5% FBS. 24 h later, the number of transmigrated cells was counted after trypsin treatment. Data were expressed as percentage of control. Cell migration data were obtained from three independent experiments.
Localization of F-Actin
F-actin was visualized by affinity-fluorescence methods. Fluorescent emissions from TRITC-phalloidin (Cytoskeleton, USA) bound to F-actin were analyzed by confocal microscopy at the same level of laser intensity for all the studied samples.
In vivo chronic hypoxic animal model
Animal experiments were carried out in accordance with the guidelines approved by the Ethics Committee of Shenzhen University. The permit number of experiment animal for this study is SCXK (Yue) 2008-0002. Chronic hypoxia-induced PAH mouse model was performed as previously described55. Lungs from PAH mice were stored at −80 °C prior to use. Rats for in vivo studies were randomized into four groups (n = 8 each): 1) a normoxic control group, 2) a hypoxic control group, 3) a hypoxic group injected with agomir control and 4) a hypoxic group injected with miR-223 agomir (Ribobio). miR-223 agomirs (30 nmol), agomir control (30 nmol), or the normal saline (NS) were injected intravenously (tail vein, 0.3 ml) at day 7 and 14. Degree of PAH were assessed by measuring right ventricular systolic pressure (RVSP) and the ratio of right to left ventricle plus septum weight [RV/(LV + S)] at day 21 as described56. After RVSP recording, whole blood were collected via right ventricular puncture, filled in 1.5 ml tubes, centrifuged for 3,000 × g for 10 min at 4 °C and serum collected for detection of miR-223 expression levels.
Western blotting
Cells were lysed with ice-cold RIPA buffer (50 mmol/LTris-HCl, pH 7.5; 150 mmol/L NaCl; 1% NP-40; 0.25% sodium deoxycholate, 1 mmol/L EDTA), supplemented with a protease inhibitor cocktail (Sigma-Aldrich). Protein concentration was determined using a conventional Coomassie Bradford protein assay kit (Bio-Rad). Equal amounts of extracts (30 μg) were then electrophoresed on sodium dodecyl sulfate polyacrylamide gel electrophoresis gel (SDS-PAGE) and electroblotted to PVDF membranes (Bio-Rad). Membranes were immersed in blocking buffer (5% degreased milk powder) for 1 h and incubated with primary antibodies overnight at 4 °C. The primary antibodies used are as follows: rabbit polyclonal anti-RhoB, -MYPT1, -MLC2, -PCNA (1:1000 dilutions, Proteintech Group), rabbit polyclonal anti-MYPT1-Phospho (thr696) (1:500 dilutions, Cell Signaling), anti-MLC2-phospho (1:200 dilutions, Cell Signaling), beta-actin (Santa Cruz Biotechnology). They were then incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson Immuno Research) and the protein bands were afterward visualized using the SuperSignal chemiluminescent detection module (Pierce) and exposed to X-ray film.
3′-UTR luciferase reporter assay
We applied TargetScan algorithm (http://www.targetscan.org) and FINDTAR3 (http://bio.sz.tsinghua.edu.cn/) to predict targets and the miRNA binding sites. The 3′-UTR of predicted target genes were PCR amplified and inserted into pmirGLO dual-luciferase vector (Promega). RhoB 3′-UTR were amplified from rat genomic DNA with primer 5′-GTC GAA TTC AGC AAG CCA CTA CTG TTG CTC CAT G-3′ (forward) and 5′-AGC TCT CGA GTC TTC TGA CAC TAT TAA GCC ACA GG-3′ (reverse). To construct mutated 3′-UTR report vector, two residues in the region that base-pairs with miRNA seeding sequences were mutated by site-directed mutagenesis. The pimers used were listed as follows: RhoB-mut1, 5′-CCC TGG GGA AGA CAT TTG CAT GAC TCT TGG GGT CGA GAG GAA GCA-3′(forward) and 5′-TGC TTC CTC TCG ACC CCA AGA GTC ATG CAA ATG TCT TCC CCA GGG-3′ (reverse); RhoB-mut2, 5′-GTG GTA CTT CTA AAT TGT CTT GTT TTG TTT TTG TTT TAT TTT TTT TAA ATA ATG ACT CAG ATG ACA AAT GGT GAA CTT ATG ATG-3′ (forward) and 5′-CAT CAT AAG TTC ACC ATT TGT CAT CTG AGT CAT TAT TTA AAA AAA ATA AAA CAA AAA CAA AAC AAG ACA ATT TAG AAG TAC CAC-3′ (reverse). The primers used for amplifying rat MLC2 3′-UTR and constructing its mutated reporter vector are listed as follows: MLC2 3′-UTR, 5′-GGA GAA TTC AGC CCC CTG ACA CCC CAG CCC CCG C-3′ (forward) and 5′- CCA CTC GAG AGA TTC TGC CCC CTC TGG GAC AGA T-3′ (reverse); MLC2-mut, 5′-AGA AGT GCG TGC CGA CGA CTC GCA GAT GTT CCC ACA-3′ (forward) and 5′-TGT GGG AAC ATC TGC GAG TCG TCG GCA CGC ACT TCT-3′ (reverse). HEK293 cells were seeded in 24-well plates and after the cells reached 80~90% confluence, each well of cells was transfected with 100 ng of 3′-UTR reporter vectors or mutated 3′-UTR reporter vectors, 900 ng of miR-223 expressing plasmid (pLVX-CMV-miR-223) with 2 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The pLVX-CMV (Clontech) without miRNA sequences was used as the control. Two days after transfection, the cells were harvested for luciferase activity assay and measured independently with a Lumat LB9508 luminometer (Berthold, Bad Wildbad, Germany). Firefly luciferase activity was normalized to renilla luciferase activity.
Rho GTPases Acthivity
Rho GTPases activity was determined by using Rho Activation Assay Biochem Kit (Cytoskeleton Inc., Denver, CO) and followed the procedures suggested by the manufacturer. Active RhoB were pulled down by rhotekin-RBD beads. Affinity-precipitated RhoB proteins were resolved by SDS-PAGE and detected by Western blot analysis using anti-RhoB antibody.
Human Serum Collection
Human serum samples were collected from healthy participants and patients with congenital heart disease associated with pulmonary arterial hypertention (CHD-PAH) at the Sun Yat-Sen Cardiovascular Hospital (Shenzhen, China) and Fuwai Hospital (Beijing, China) in accordance with the guidelines approved by the Ethics Committee of Shenzhen University and the associated hospitals. The blood samples were allowed to stand at room temperature for 1 h and then centrifuged at 3,000 × g for 10 min at 4 °C. The supernatants were stored at −80 °C. The study was approved by both the ethics committees of the Sun Yat-Sen Cardiovascular Hospital and Fuwai Hospital. All subjects who participated in the study provided written informed consent.
Statistical analysis
All data shown are mean values of at least three experiments, each performed in triplicate, with standard errors (SE). The differences between only two groups were analyzed using the double-sided Student’s t test and with three or more groups were analyzed using ANOVA test. A p value less than 0.05 was considered significant. The significant differences (p < 0.05 or p < 0.01) are indicated in the figures by asterisk.
Additional Information
How to cite this article: Zeng, Y. et al. MicroRNA-223 Attenuates Hypoxia-induced Vascular Remodeling by Targeting RhoB/MLC2 in Pulmonary Arterial Smooth Muscle Cells. Sci. Rep. 6, 24900; doi: 10.1038/srep24900 (2016).
References
Archer, S. L., Weir, E. K. & Wilkins, M. R. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 121, 2045–66 (2010).
Humbert, M. et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43, 13S–24S (2004).
Crosswhite, P. & Sun, Z. Molecular mechanisms of pulmonary arterial remodeling. Mol Med 20, 191–201 (2014).
Heasman, S. J. & Ridley, A. J. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9, 690–701 (2008).
Wojciak-Stothard, B. et al. Role of RhoB in the regulation of pulmonary endothelial and smooth muscle cell responses to hypoxia. Circ Res 110, 1423–34 (2012).
Kajimoto, H. et al. Oxygen activates the Rho/Rho-kinase pathway and induces RhoB and ROCK-1 expression in human and rabbit ductus arteriosus by increasing mitochondria-derived reactive oxygen species: a newly recognized mechanism for sustaining ductal constriction. Circulation 115, 1777–88 (2007).
Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism and function. Cell 116, 281–97 (2004).
Dang, L. T., Lawson, N. D. & Fish, J. E. MicroRNA control of vascular endothelial growth factor signaling output during vascular development. Arterioscler Thromb Vasc Biol 33, 193–200 (2013).
Davis, B. N., Hilyard, A. C., Lagna, G. & Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 454, 56–61 (2008).
Sarkar, J. et al. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol 299, L861–71 (2010).
Courboulin, A. et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 208, 535–48 (2011).
Rangrez, A. Y., Massy, Z. A., Metzinger-Le Meuth, V. & Metzinger, L. miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet 4, 197–205 (2011).
Cordes, K. R. et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460, 705–10 (2009).
Kang, K. et al. MicroRNA-124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. J Biol Chem 288, 25414–27 (2013).
Wang, D. et al. MicroRNA-124 controls the proliferative, migratory and inflammatory phenotype of pulmonary vascular fibroblasts. Circ Res 114, 67–78 (2014).
Tang, H., Ayon, R. J. & Yuan, J. X. New insights into the pathology of pulmonary hypertension: implication of the miR-210/ISCU1/2/Fe-S axis. EMBO Mol Med 7, 689–91 (2015).
Chen, T. et al. Loss of microRNA-17 approximately 92 in smooth muscle cells attenuates experimental pulmonary hypertension via induction of PDZ and LIM domain 5. Am J Respir Crit Care Med 191, 678–92 (2015).
Brock, M. et al. AntagomiR directed against miR-20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia-induced pulmonary hypertension. Eur Heart J 35, 3203–11 (2014).
Parikh, V. N. et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 125, 1520–32 (2012).
Lu, H., Buchan, R. J. & Cook, S. A. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc Res 86, 410–20 (2010).
Sun, W. et al. miR-223 and miR-142 attenuate hematopoietic cell proliferation and miR-223 positively regulates miR-142 through LMO2 isoforms and CEBP-beta. Cell Res 20, 1158–69 (2010).
Eyholzer, M. et al. Complexity of miR-223 regulation by CEBPA in human AML. Leuk Res 34, 672–6 (2010).
Wong, Q. W. et al. MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology 135, 257–69 (2008).
Laios, A. et al. Potential role of miR-9 and miR-223 in recurrent ovarian cancer. Mol Cancer 7, 35 (2008).
Harraz, M. M., Eacker, S. M., Wang, X., Dawson, T. M. & Dawson, V. L. MicroRNA-223 is neuroprotective by targeting glutamate receptors. Proc Natl Acad Sci USA 109, 18962–7 (2012).
Greco, S. et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J 23, 3335–46 (2009).
Shi, L. et al. MicroRNA-223 antagonizes angiogenesis by targeting beta1 integrin and preventing growth factor signaling in endothelial cells. Circ Res 113, 1320–30 (2013).
Pietra, G. G. et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol 43, 25S–32S (2004).
Chen, B., Calvert, A. E., Cui, H. & Nelin, L. D. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol 297, L1151–9 (2009).
Totsukawa, G. et al. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol 150, 797–806 (2000).
Singh, D. K., Sarkar, J., Raghavan, A., Reddy, S. P. & Raj, J. U. Hypoxia modulates the expression of leucine zipper-positive MYPT1 and its interaction with protein kinase G and Rho kinases in pulmonary arterial smooth muscle cells. Pulm Circ 1, 487–98 (2011).
Di Stefano, V., Zaccagnini, G., Capogrossi, M. C. & Martelli, F. microRNAs as peripheral blood biomarkers of cardiovascular disease. Vascul Pharmacol 55, 111–8 (2011).
Wang, G. K. et al. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J 31, 659–66 (2010).
Wei, C. et al. Circulating miRNAs as potential marker for pulmonary hypertension. PLos One 8, e64396 (2013).
Evans, A. M., Hardie, D. G., Peers, C. & Mahmoud, A. Hypoxic pulmonary vasoconstriction: mechanisms of oxygen-sensing. Curr Opin Anaesthesiol 24, 13–20 (2011).
Hale, A. E., White, K. & Chan, S. Y. Hypoxamirs in Pulmonary Hypertension: Breathing New Life into Pulmonary Vascular Research. Cardiovasc Diagn Ther 2, 200–212 (2012).
Caruso, P. et al. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol 30, 716–23 (2010).
Rangrez, A. Y. et al. Inorganic phosphate accelerates the migration of vascular smooth muscle cells: evidence for the involvement of miR-223. PLos One 7, e47807 (2012).
Meloche, J. et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 129, 786–97 (2014).
Meloche, J. et al. MiR-223 Reverses Experimental Pulmonary Arterial Hypertension. Am J Physiol Cell Physiol. ajpcell 00149 2015 (2015).
Sun, G., Li, H. & Rossi, J. J. Sequence context outside the target region influences the effectiveness of miR-223 target sites in the RhoB 3′UTR. Nucleic Acids Res 38, 239–52 (2010).
Raman, N. & Atkinson, S. J. Rho controls actin cytoskeletal assembly in renal epithelial cells during ATP depletion and recovery. Am J Physiol 276, C1312–24 (1999).
Lu, Q., Longo, F. M., Zhou, H., Massa, S. M. & Chen, Y. H. Signaling through Rho GTPase pathway as viable drug target. Curr Med Chem 16, 1355–65 (2009).
Ji, X. et al. Plasma miR-208 as a biomarker of myocardial injury. Clin Chem 55, 1944–9 (2009).
Tanaka, M. et al. Down-regulation of miR-92 in human plasma is a novel marker for acute leukemia patients. PLos One 4, e5532 (2009).
Chen, X., Liang, H., Zhang, J., Zen, K. & Zhang, C. Y. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol 22, 125–32 (2012).
Mitchell, P. S. et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA 105, 10513–8 (2008).
Zampetaki, A. et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res 107, 810–7 (2010).
Zampetaki, A. et al. Prospective study on circulating MicroRNAs and risk of myocardial infarction. J Am Coll Cardiol 60, 290–9 (2012).
Zhang, Y. Y. et al. Decreased circulating microRNA-223 level predicts high on-treatment platelet reactivity in patients with troponin-negative non-ST elevation acute coronary syndrome. J Thromb Thrombolysis 38, 65–72 (2013).
de Torres, J. P. et al. Gender differences in plasma biomarker levels in a cohort of COPD patients: a pilot study. PLos One 6, e16021 (2011).
Ma, K. et al. Human lung microRNA profiling in pulmonary arterial hypertension secondary to congenital heart defect. Pediatr Pulmonol 50, 1214–23 (2015).
Kang, K. et al. A novel real-time PCR assay of microRNAs using S-Poly(T), a specific oligo(dT) reverse transcription primer with excellent sensitivity and specificity. PLoS One 7, e48536 (2012).
Niu, Y. et al. An improved method for detecting circulating microRNAs with S-Poly(T) Plus real-time PCR. Sci Rep 5, 15100 (2015).
Gou, D. et al. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am J Physiol Lung Cell Mol Physiol 303, L682–91 (2012).
Shimoda, L. A. & Semenza, G. L. Functional analysis of the role of hypoxia-inducible factor 1 in the pathogenesis of hypoxic pulmonary hypertension. Methods Enzymol 381, 121–9 (2004).
Acknowledgements
We thank Dr. Xuemei Chen for comments on this manuscript. This study was supported by National Natural Science Foundation of China [81170047, 81370151 and 81570046 to D.G., 81173112, 81470246, 81170052 and 81220108001 to J.W. and 31571199 to K.K.]; National Basic Research Program of China 973 Program [2012CB124701 to D.G.]; Shenzhen Municipal Basic Research Program [JC201006010725A to D.G.]; Interdisciplinary Innovation Team Project of Shenzhen University; Shenzhen Overseas High-Level Talents Innovation Program [YFZZ20111009 to D.G.]; Shenzhen High-tech Development Project [CXZZ20140828163951592 to D.G.]; Shenzhen Municipal Basic Research Program [JCYJ20130329120507746 to K.K.]; and Shenzhen University Foundation [201562 to K.K.]. Funding for open access charge: National Natural Science Foundation of China.
Author information
Authors and Affiliations
Contributions
D.G. and K.K. designed experiments and analyzed data; X.Z., K.K., R.R., J.C., Z.W., W.L., Y.C. and J.Z. performed experiments; Z.W., J.H. and Y.Z. collected the clinical samples; Y.Z. and D.G. wrote the manuscript; R.R., U.R. and J.Q. edited the manuscripts; D.G., U.R. and J.W. supervised the project. All authors read and approved the final version of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zeng, Y., Zhang, X., Kang, K. et al. MicroRNA-223 Attenuates Hypoxia-induced Vascular Remodeling by Targeting RhoB/MLC2 in Pulmonary Arterial Smooth Muscle Cells. Sci Rep 6, 24900 (2016). https://doi.org/10.1038/srep24900
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24900
This article is cited by
-
Discovering novel clues of natural selection on four worldwide goat breeds
Scientific Reports (2023)
-
T18/S19 diphosphorylation of myosin regulatory light chain impairs pulmonary artery relaxation in monocrotaline-induced pulmonary hypertensive rats
Pflügers Archiv - European Journal of Physiology (2023)
-
Signatures of selection reveal candidate genes involved in economic traits and cold acclimation in five Swedish cattle breeds
Genetics Selection Evolution (2020)
-
An improved Tet-on system in microRNA overexpression and CRISPR/Cas9-mediated gene editing
Journal of Animal Science and Biotechnology (2019)
-
Genomic signatures of high-altitude adaptation in Ethiopian sheep populations
Genes & Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
