
Abstract
Xanthohumol (XN) is a natural anticancer compound that inhibits the proliferation of oestrogen receptor-α (ERα)-positive breast cancer cells. However, the precise mechanism of the antitumour effects of XN on oestrogen (E2)-dependent cell growth and especially its direct target molecule(s), remain(s) largely unknown. Here, we focus on whether XN directly binds to the tumour suppressor protein prohibitin 2 (PHB2), forming a novel natural antitumour compound targeting the BIG3-PHB2 complex and acting as a pivotal modulator of E2/ERα signalling in breast cancer cells. XN treatment effectively prevented the BIG3-PHB2 interaction, thereby releasing PHB2 to directly bind to both nuclear- and cytoplasmic ERα. This event led to the complete suppression of the E2-signalling pathways and ERα-positive breast cancer cell growth both in vitro and in vivo, but did not suppress the growth of normal mammary epithelial cells. Our findings suggest that XN may be a promising natural compound to suppress the growth of luminal-type breast cancer.
Similar content being viewed by others

Introduction
Oestrogen-receptor-α (ERα) plays a pivotal role in the development and progression of breast cancer. The current endocrine therapies for breast cancer are mainly based on targeting ERα signalling using selective ERα modulators (e.g., tamoxifen and raloxifene), ERα downregulators (e.g., fulvestrant) and aromatase inhibitor (AI)1,2,3. Of these treatments, tamoxifen, which inhibits breast cancer cell growth through the competitive binding of ERα, is a standard treatment offered to patients with ERα-positive breast cancer. However, tamoxifen treatment often fails and patients succumb to recurrent, endocrine-resistant tumours4,5. Moreover, AI, which blocks oestrogen (E2) synthesis, provides substantial clinical benefits, such as good efficacy, a significant increase in disease-free survival and a prolonged time to disease recurrence in postmenopausal women with ERα-positive breast cancer, compared with tamoxifen treatment. Nevertheless, some patients who have undergone AI treatment still relapse6,7. The precise molecular events that determine alterations in the effectiveness of these endocrine therapies remain unknown.
We previously reported that the oncoprotein brefeldin A-inhibited guanine nucleotide-exchange protein 3 (BIG3) and tumour suppressor prohibitin 2 (PHB2) complex play a pivotal role in E2 signalling modulation in ERα-positive breast cancer8,9. Namely, BIG3 binds PHB2, thereby inhibiting the E2-dependent suppressive ability of PHB2 and resulting in constitutive ERα activation. Considering these findings, strategies utilising the tumour suppressive activity of PHB2 upon its release from BIG3 by inhibitors of protein-protein interaction (PPI) may represent novel therapies for breast cancer, although PPI has been difficult to target with small molecules or synthetic peptide inhibitors. Indeed, we further demonstrated that a dominant-negative peptide, ERAP, which specifically disrupts the BIG3-PHB2 interaction, leads to the inhibition of multiple ERα-signalling pathways driving the growth of breast cancer by reactivating PHB2 tumour suppressive activity. However, because this peptide is difficult to use in clinical practice due to its limited stability, it is necessary to identify alternative long-term stable antagonistic compounds targeting the BIG3-PHB2 interaction.
Xanthohumol (XN: (2E)-1-[2,4-dihydroxy-6-methoxy-3-(3-methyl-2-buten-1-yl) phenyl]-3-(4-hydroxyphenyl)-2-propen-1-one), a prenylated chalcone present in hops (Humulus lupulus L.), has been shown to inhibit the growth of a wide variety of human cancer cell lines, including breast, ovarian, liver, colon and prostate cancer cell lines10,11. Most notably, several studies have reported that XN inhibits the proliferation of the breast cancer cell lines MCF-7 and SK-BR-3 both in vitro and in vivo12,13,14,15,16. However, these studies demonstrated an inhibition of cell proliferation in ERα-positive breast cancer cells in the absence of E2 stimulation or ERα-negative breast cancer cells. In fact, the precise mechanism of the antitumour effect of XN on E2-dependent growth in luminal-type breast cancer cells remains largely unknown. To clarify the precise mechanism of the antitumour effects of XN against breast cancer, we focused on the evidence that XN directly binds to the PHB2 protein17. Considering these and our previous findings, we hypothesised that the inhibition of the BIG3-PHB2 interaction by the direct binding of XN to PHB2 may lead to the suppression of constitutive ERα activation in breast cancer cells. Here, we found that XN inhibits E2/ERα signalling by reactivating the tumour-suppressive ability of PHB2 and its antitumour effect is more effective than that of ERAP.
Results
Xanthohumol inhibits the BIG3-PHB2 interaction and mediates the repression of multiple E2-induced activation events
Previous reports show that XN inhibits the proliferation of ERα-positive breast cancer cells12,13,14,15,16 and specifically binds to the prohibitin proteins PHB1 and PHB217. Moreover, we recently reported that BIG3-PHB2 complex formation plays a crucial role in ERα-positive breast cancer cell growth8,9. Considering these findings, we attempted to investigate the possibility of XN as an inhibitor targeting the BIG3-PHB2 interaction. Co-immunoprecipitation experiments revealed that XN dose-dependently inhibited the complex formation of endogenous BIG3 and PHB2 in the ERα-positive breast cancer cell lines MCF-7 and KPL-3C, which highly express both BIG3 and PHB2 (Fig. 1a and Supplementary Fig. S1a). Similar results were obtained with ERAP treatment, a dominant-negative peptide inhibiting the BIG3-PHB2 interaction by its direct binding to PHB2 that we previously developed9 (Fig. 1a and Supplementary Fig. S1a). We also demonstrated that XN directly bound to recombinant PHB2 protein in vitro (Supplementary Fig. S1b). In addition, we found that XN inhibited the in vitro PHB2-ERAP interaction in a dose-dependent manner (Supplementary Fig. S1c), suggesting the possibility that XN caused the specific inhibition of BIG3-PHB2 complex formation by its direct binding to PHB2.

Xanthohumol inhibits the BIG3-PHB2 interaction and mediates the repression of multiple E2-induced activation events.
(a)The inhibitory effects of XN treatment on BIG3-PHB2 interactions were evaluated in MCF-7 cells; ERAP was used as a positive control for the inhibition of the BIG3-PHB2 interaction. The blots were cropped and the full-length blots were included in the supplementary information. (b) The interaction of ERα with PHB2 released by XN treatment in the cytoplasmic and nuclear fractions was evaluated. α/β-Tubulin was used as loading controls for the cytoplasmic fraction. The blots were cropped and the full-length blots were included in the supplementary information. (c) Representative immunofluorescence images of the subcellular localisation of PHB2 are shown. (d) The inhibitory actions of XN on ERα transcriptional activity were evaluated using luciferase assays. The data represent the mean ± SE of three independent experiments (** P < 0.01, *** P < 0.001 in two-sided Student's t-test). (e) Immunoblot analyses were performed to evaluate the inhibitory effects of XN on E2-induced Akt (S473), MAPK (T202/Y204) and ERα (S104/S106, S118, S167, S305 and Y537) activities in MCF-7 cells. The blots were cropped and the full-length blots were included in the supplementary information.
We next investigated the subcellular distribution of endogenous PHB2 in ERα-positive breast cancer cells following XN treatment. The results showed that in the presence of E2, treatment with XN led to a decrease in cytoplasmic PHB2, thereby substantially increasing the amount of nuclear PHB2 (Fig. 1b, WCL). Furthermore, co-immunoprecipitation experiments with an anti-ERα antibody indicated that the PHB2 released from BIG3 by XN treatment interacts with cytoplasmic and nuclear ERα in a dose-dependent manner (Fig. 1b, IP: ERα).
Next, we sought to elucidate the effect of XN on nuclear ERα function as a transcriptional factor. After validating E2-dependent PHB2 nuclear translocation by XN treatment using immunocytochemical approaches with MCF-7 and KPL-3C cells (Fig. 1c and Supplementary Fig. S1d), we analysed ERα transcriptional activity by luciferase reporter assays with oestrogen-responsive element (ERE) reporters. XN significantly inhibited E2-induced ERα transcriptional activity in a dose-dependent manner in MCF-7 and KPL-3C cells (minimum effective dose, 5 μM; Fig. 1d and Supplementary Fig. S1e).
We previously demonstrated that inhibiting the BIG3-PHB2 interaction interfered with E2-induced, non-genomic ERα activation pathways9. We next examined the effects of XN on the phosphorylation status of Akt and MAPK, which are the downstream signalling molecules of the non-genomic pathways18,19,20,21. As expected, Akt (S473) and p42/44 MAPK (T202/Y204) phosphorylation levels were increased after 24 h of E2 stimulation in MCF-7 cells, whereas 30 μM XN treatment clearly abrogated the E2-induced phosphorylation levels of both proteins as well as that induced by ERAP treatment (Fig. 1e). In addition, 30 μM XN treatment remarkably reduced the phosphorylation levels at all five sites (S104/S106, S118, S167, S305 and Y537) within ERα, which are involved in ERα transcriptional activity, DNA-binding, co-activator binding, protein stability and cell proliferation in ERα-positive breast cancer cells22,23,24,25,26,27,28,29 (Fig. 1e). The amplitude of the suppression was comparable with that of the positive control ERAP. These results suggest that XN regulates ERα signalling, which is important for E2-dependent cell proliferation in ERα-positive breast cancer cells. Collectively, these findings indicate that XN treatment led to intrinsic PHB2 binding to ERα, thereby repressing multiple E2-induced activation events, including genomic and non-genomic ERα-activation and ERα phosphorylation in breast cancer cells.
XN specifically suppresses the E2-dependent cell growth of breast cancer
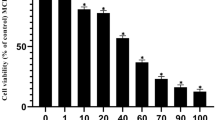
To examine the effect of XN on breast cancer cell proliferation, we measured the E2-dependent cell growth of ERα-positive breast cancer cell lines, MCF-7 or KPL-3C, with an MTT assay at 24 h after treatment with XN. The results showed that XN treatment significantly reduced the E2-stimulated cell growth in a dose-dependent manner (IC50 = 7.3 μM and 8.2 μM in the MCF-7 and KPL-3C cells, respectively; Fig. 2a). By contrast, 50 μM XN remarkably inhibited the cell proliferation of both MCF-7 and KPL-3C cells, indicating the inhibition of E2-independent cell growth (Fig. 2a). In fact, similar results were obtained with MCF-7 cell growth in the absence of E2 (Supplementary Fig. S2a), suggesting that 50 μM XN inhibits cell growth by independent mechanisms targeting the BIG3-PHB2 interaction. By contrast, compared with MCF-7 or KPL-3C cells, 30 μM XN had little effect on the growth of normal mammary epithelial MCF-10A cells that do not express ERα or BIG3 (Fig. 2b and Supplementary Fig. S2b). Accordingly, it must be noted that 50 μM XN treatment induced a non-specific inhibitory effect on the growth of MCF-10A cells. Furthermore, we also demonstrated that 30 μM XN treatment had no inhibitory effects on E2-induced growth of siRNA-mediated PHB2-depleted cells (Supplementary Fig. S2c), indicating that XN suppresses E2-induced cell growth through its specific inhibition of BIG3-PHB2 interaction. These results indicate that 30 μM XN specifically and completely inhibited multiple aspects of the E2/ERα-signalling network via the tumour-suppressive ability of endogenous PHB2 released from BIG3, resulting in complete suppression of E2-dependent breast cancer cell growth.

Xanthohumol suppresses E2-dependent cell growth with long-term stability.
(a,b) An MTT assay was performed to evaluate the inhibitory effect of XN on the E2-dependent growth of the BIG3-positive cell lines MCF-7 (a, left) or KPL-3C (a, right) and the growth of the BIG3-negative mammary epithelial cell line MCF-10A (b) for 24 h. The data represent the mean ± SE of three independent experiments (* P < 0.05, ** P < 0.01, *** P < 0.001 in two-sided Student's t-test). (c,d) The duration of the inhibitory effects of XN on cell growth (c) and ERα-target gene expression (d) were measured in MCF-7 cells for the times indicated. The results were expressed as the fold increase over untreated cells at each time (c) and at 0 h (d, set at 1.0). The data represent the mean ± SE of three independent experiments (* P < 0.05, ** P < 0.01 in two-sided Student's t-test).
We next investigated the duration of the inhibitory effect of XN by an MTT assay. In contrast with ERAP, the inhibitory effect of XN on the growth of the MCF-7 and KPL-3C breast cancer cells was maintained for 96 h after XN treatment without affecting cell morphology (Fig. 2c and Supplementary Figs. S2d–g). In addition, XN also significantly suppressed E2-induced expression of the ERα-target genes TFF1 and CCND1 for 96 h after XN treatment (Fig. 2d). Taken together, our findings strongly suggest that the inhibitory effect of XN on the responsiveness of ERα-positive breast cancer cells has much higher stability than that of ERAP.
Next, we examined the effects of XN on the cell cycle distribution of MCF-7 cells by flow cytometry and microscopy analyses. The population of cells in the G2/M phase increased after a 24 h E2 stimulation, whereas the population in the G1 phase, but not sub-G1 population, increased after 30 μM XN treatment, indicating that XN suppressed cell growth by inducing a G1 arrest and caused no cell phenotypic alterations, similar to ERAP (Fig. 3a,b; ref. 9). By contrast, high dose of XN treatment causes cell phenotypic alterations regardless of E2 stimulation (Supplementary Fig. S3). Similarly, XN has been reported to induce apoptosis in several cancer cell lines, including leukemia, prostate cancer and hepatoma cell lines30. We previously reported that XN acts as an autophagy modulator by directly binding to the N-domain of valosin-containing protein (VCP), which is essential for autophagosome-lysosome fusion and the formation of autolysosomes in cancer cells17. Therefore, we wished to clarify the effect of XN on VCP function in ERα-positive breast cancer cells. We first examined VCP expression in various cell lines and found high expression of VCP in breast cancer cell lines as well as in colon and lung cancer cell lines and a mammary epithelial cell line (Fig. 3c). Next, because XN was also reported to induce endoplasmic reticulum (ER) stress via inducing the loss of VCP-mediated ER-associated degradation (ERAD) activity17, we examined the effect of XN treatment on the expression of CHOP and GRP78, ER stress-response genes. The results showed that 50 μM XN treatment, but not 30 μM XN, caused remarkable up-regulation of both genes at transcriptional levels regardless of E2 stimulation (Fig. 3d). Furthermore, we examined whether the inhibitory effect of 30 μM XN treatment is due to its loss of VCP function. The results showed that 30 μM XN treatment could cause a complete suppression of E2-induced growth of VCP-depleted cells, suggesting that the inhibitory effect of 30 μM XN is not dependent on the loss of VCP function although 50 μM XN treatment might cause apoptosis through its inhibition of VCP function (Fig. 3e). Taken together, our data clearly demonstrate that XN suppresses E2-induced cell growth via specific disruption of the BIG3-PHB2 interaction regardless of VCP function.

Xanthohumol suppresses E2-dependent cell growth independently of apoptosis induction and VCP function.
(a) Flow cytometric analyses were performed to evaluate the effect of XN treatment on the cell cycle. MCF-7 cells were treated for 24 h with E2 and/or XN or ERAP. (b) Representative cell morphologies of MCF-7 or MCF-10A cell following XN treatment are shown. (c) Expression patterns of VCP, BIG3 and PHB2 in breast cancer and normal epithelial cell lines. Colorectal cancer cell lines (HCT116 and SW480) and an epidermoid carcinoma cell line (A431) were used as positive controls for VCP expression. β-Actin served as a quantitative internal control. The blots were cropped and the full-length blots were included in the supplementary information. (d) The CHOP and GRP78 expression levels following XN treatment were evaluated using real-time PCR. The data represent the mean ± SE of three independent experiments (* P < 0.05, ** P < 0.01, *** P < 0.001 in two-sided Student's t-test). (e) An MTT assay was performed to evaluate the inhibitory effect of XN on the E2-dependent growth of VCP-depleted cells. The data represent the mean ± SE of three independent experiments (** P < 0.01, *** P < 0.001 in two-sided Student's t-test).
XN inhibits ERα-positive breast cancer tumour growth in a xenograft mouse model
To determine the antitumour activity of XN in vivo, KPL-3C orthotopic breast cancer xenografts were developed in nude mice. Once the tumours were fully established, various concentrations of XN (0.3 and 1.0 mg kg−1), ERAP (14 mg kg−1), or vehicle alone were administered daily by intraperitoneal injection for 36 days. The animals also received daily treatments of E2 (6 µg per day). Daily E2 treatment induced the time-dependent growth of KPL-3C tumours, whereas XN treatment caused a significant inhibition of E2-induced tumour growth comparable with that observed after ERAP treatment (Fig. 4a and Supplementary Fig. S4a). No toxicity or significant body weight changes were observed throughout these experiments (Supplementary Fig. S4b). To clarify the mechanisms of the in vivo antitumour effect of XN, we first examined its effect on the BIG3-PHB2 interaction. Co-immunoprecipitation experiments in each tumour revealed that 1.0 mg kg−1 and 0.3 mg kg−1 XN treatment effectively inhibited the formation of the endogenous BIG3-PHB2 complex, resulting in nuclear translocation of PHB2 in tumours (Fig. 4b). Substantially increased nuclear PHB2 staining was observed in tumours treated with XN in a dose-dependent manner by immunohistochemistry (Fig. 4c). We next examined the effects of XN on the activation of the non-genomic ERα signalling pathway. As expected, considerable suppression of Akt and MAPK phosphorylation was observed in tumours treated with XN (Fig. 4d), demonstrating that XN had potent in vivo antitumour activity. Furthermore, to evaluate the effect of XN treatment on the cell cycle in tumours, we investigated the expression of the proliferative markers, Ki67 and PCNA by immunohistochemistry31. Ki67 is known to be expressed in all phases of the active cell cycle (G1, S, G2 and M phase) and PCNA in most abundant during the S phase32. As shown in Fig. 4e, Ki67 was clearly detected in every tumour, whereas PCNA was not detected in tumours treated with 1.0 mg kg−1 XN or 14 mg kg−1 ERAP, indicating that XN treatment suppresses tumour growth by inducing G1-arrest. Collectively, these results suggest that XN acts as an effective therapeutic agent against ERα-positive breast cancer by inducing G1-arrest via targeting the BIG3-PHB2 interaction.

Xanthohumol has in vivo anti-tumour efficacy in xenograft models of human ERα-positive breast cancer.
(a) XN inhibits tumour growth in a human breast cancer KPL-3C xenograft mouse model. The tumour size represents the mean ± SE of each group (n = 5). (* P < 0.05, ** P < 0.01 in two-sided Student's t-test). (b) The inhibitory effects of XN treatment on the BIG3-PHB2 interactions were evaluated in tumours. The blots were cropped and the full-length blots were included in the supplementary information. (c) Statistical analyses of the intensity (upper) and representative immunohistochemical staining (lower) of nuclear PHB2 protein in tumours. The data represent the mean ± SE of five different points (* P < 0.05, ** P < 0.01 in two-sided Student's t-test). (d) Immunoblot analyses were performed to evaluate the effects of XN on the phosphorylation levels of Akt and MAPK proteins in tumours. The blots were cropped and the full-length blots were included in the supplementary information. (e) Representative immunohistochemical staining of nuclear Ki67 (upper) and PCNA (lower) in tumours (left) and statistical analyses of the nuclear PCNA intensity (right).
Discussion
Previous studies have shown that the BIG3-PHB2 complex plays a critical role in promoting breast cancer cell growth8,9 and strategies capable of inhibiting this interaction may represent novel therapies for breast cancer. In this study, we demonstrated that a natural antitumour compound, XN, which binds to endogenous PHB2, prevented the BIG3-PHB2 interaction, thereby releasing PHB2 to inhibit both nuclear- and membrane-associated ERα activation. These data are in accordance with our previous observations that a dominant-negative peptide, ERAP targeting the BIG3-PHB2 interaction suppressed the growth of ERα-positive breast cancer cells9. Collectively, therapeutic strategies utilising the reactivation of PHB2 upon its PPI inhibitor-mediated release from BIG3 appeared to effectively treat luminal-type breast cancer cells, especially those with endocrine resistance. However, because the inhibitory effect of ERAP is maintained for only 24 h9, it is difficult to use in clinical practice. Therefore, we tested the inhibitory effect of XN, which directly binds to the PHB2 protein, as a selective antagonistic compound targeting the BIG3-PHB2 interaction and found that the inhibitory effect of XN demonstrated long-term stability was maintained for up to 96 h (Figs. 2c and d). Moreover, no toxicity or significant body weight loss by daily intraperitoneal injection with effective doses for 36 days were observed. In addition, previous studies have shown that long-term treatment with XN causes no adverse effects on female mouse reproduction32. From a therapeutic perspective, XN targeting of the BIG3-PHB2 interaction provides excellent therapeutic indices with minimal off-target effects, thereby leading to better quality-of-life for patients with breast cancer.
Another interesting finding of this study is that 30 μM XN specifically inhibited multiple E2-induced cell growth pathways in breast cancer by targeting of the BIG3-PHB2 interaction despite the fact that XN is known to exert a broad spectrum of antitumour actions, such as apoptosis induction10,12,29,33 and NF-κB inhibition34,35. In particular, we previously reported that XN bound directly to the N domain of VCP, thereby inducing endoplasmic reticulum (ER) stress via the loss of VCP-mediated ER-associated degradation (ERAD) activity17. Here, we here examined the effect of XN on ER stress induction and demonstrated that 50 μM XN, but not 30 μM XN, caused remarkable up-regulation of the ER stress-response genes CHOP and GRP78 in breast cancer cells regardless of E2 stimulation (Fig. 3d). These findings suggest that the antitumour effect of 30 μM XN is dependent on the inhibition of the BIG3-PHB2 interaction, but not on the loss of VCP function. However, high-dose (50 μM) XN treatment might cause loss of VCP function, thereby leading to the accumulation of unfolded proteins and resulting in ER-stress in cancer cells36,37. Furthermore, we observed that 30 μM XN resulted in complete suppression of the E2-induced cell growth in VCP-depleted cells (Fig. 3e). Taken together, XN (30 μM) treatment appears to be a natural antitumour compound targeting BIG3-PHB2 complex formation in breast cancer cells, although it will be necessary to clarify the effect of XN treatment on other binding partners involved in NF-κB signalling and apoptosis-related proteins.
Furthermore, we recently demonstrated that the combination of tamoxifen and ERAP induced more potent anti-tumour activity in vivo and in vitro compared with either treatment alone9. Thus, a combination of the current endocrine therapies and BIG3-PHB2 interaction inhibitors, such as XN or ERAP, may lead to more effective combined effects on the intrinsic and acquired endocrine resistance of breast cancer through the different modes of actions of these drugs. In conclusion, our findings show that the selective BIG3-PHB2 interaction inhibitor XN could completely suppress E2-dependent ERα activation and had growth-suppressive effects on breast cancer cells both in vitro and in vivo. These findings suggest that this BIG3-PHB2 interaction inhibitor may have great potential in the treatment of luminal type breast cancers, especially those with endocrine resistance.
Methods
Ethical statement
All experiments in this study were conducted according to protocols reviewed and approved by the Committee for Safe Handling of Living Modified Organisms (Permission number 26–40) and the Institutional Animal Care and Use Committee (Permission number 13135) in the University of Tokushima.
Cell lines and culture conditions
Human breast cancer cell lines (MCF-7, ZR-75-1, BT474, SK-BR-3, MDA-MB-453, MDA-MB-231), a mammary epitherial cell line (MCF-10A), colorectal cancer cell lines (HCT116, SW480) and epidermoid carcinoma cell line (A431) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The KPL-3C cells were established, characterized and kindly provided by Dr. Jun-ichi Kurebayashi (Kawasaki Medical School)38. All of the cell lines were cultured under the respective depositor's recommendations. The MCF-7 cells were seeded in 48-well plates (2 × 104 cells mL−1), 24-well plates (1 × 105 cells mL−1), 6-well plates (3 × 105 cells 2 mL−1), or 10-cm dishes (2 × 106 cells 10 mL−1) in MEM (Life Technologies, Rockville, MD, USA) supplemented with 10% FBS (Nichirei Biosciences, Tokyo, Japan), 1% antibiotic/antimycotic solution (Life Technologies), 0.1 mM NEAA (Life Technologies), 1 mM sodium pyruvate (Life Technologies) and 10 μg mL−1 insulin (Sigma, St. Louis, MO, USA). The cells were maintained at 37°C with 5% CO2. The next day, the medium was changed to phenol red-free DMEM/F12 (Life Technologies), supplemented with FBS, antibiotic/antimycotic solution, NEAA, sodium pyruvate and insulin. After 24 h, the cells were treated with 10 nM 17-oestradiol (E2, Sigma) ± xanthohumol or peptides (e.g., ERAP).
Antibodies and immunoblot analyses
The anti-BIG3 monoclonal antibody was generated by Sigma-Aldrich (Tokyo, Japan). Briefly, a rat was immunised with purified His-tagged human BIG3 protein (459-572 aa). The iliac lymph nodes were collected and fused with myeloma cells, resulting in the formation of a hybridoma. Immunoblot analyses were performed as described previously9. After SDS-PAGE, the membranes blotted with proteins were blocked with 4% BlockAce solution (Dainippon Pharmaceutical, Osaka, Japan) for 1 h and then incubated with antibodies against the following proteins: BIG3 (1:1,000); PHB2 (1:1,000, Abcam, Cambridge, UK); ERα (SP-1, 1:500, Thermo Fisher Scientific, Fremont, CA, USA); Akt, phospho-Akt (S473) (587F11, 1:1,000), p44/42 MAPK, phospho-p44/42 MAPK (T202/Y204) (1:1,000), VCP (valosin-containing protein) (1:500) and α/β-tubulin (1:1,000) (Cell Signaling Technology, Danvers, MA, USA); β-actin (AC-15; 1:5,000) and LMNB1 (1:100; Sigma) using standard procedures. All of the experiments were performed in triplicate at a minimum.
Immunoprecipitation
Immunoprecipitation analysis was performed as described previously9. The cell lysates were pre-cleared with normal IgG and rec-Protein G Sepharose 4B (Life Technologies) at 4°C for 3 h. Then, the supernatants were incubated with antibodies against BIG3 (5 μg) and ERα (5 μg) at 4°C for 12 h. Next, the antigen−antibody complexes were precipitated with rec-Protein G Sepharose 4B at 4°C for 1 h. The immunoprecipitated protein complexes were washed three times with lysis buffer. Subsequent SDS-PAGE and immunoblot analyses were performed as described above.
Direct binding of XN with recombinant PHB2 protein
Direct binding of XN to recombinant PHB2 protein was evaluated by XN-beads pull-down assay. The 3.6 μg of 6 x His-tagged recombinant PHB2 (His-PHB2) were incubated with 3 μL control-beads or XN-beads together with 1 mg mL−1 BSA for 4 h. Then, the bound fractions were immunoblotted with anti-6xHis antibody (Takara, Shiga, Japan).
Ni-resin agarose pull-down assay
Binding inhibition of PHB2-ERAP interaction by XN was assayed by Ni-resin agarose pull-down. The binding assay was performed by mixing 0.27 nmole 6 x His-tagged recombinant PHB2, 0.27 nmole HA-ERAP and 0.5 to 3 times moles of XN for 1 h at 4°C, followed by capturing Ni-NTA agarose (Qiagen, Hilden, Germany) for 2 h at 4°C. The bound fractions were washed with 5 mM imidazole five times and further eluted by SDS-sample buffer for immunoblot analyses.
Inhibition of the BIG3-PHB2 interaction by xanthohumol
The XN was isolated and purified as described previously17. XN was dissolved in dimethylsulphoxide (DMSO) at a concentration of 100 mmol L−1 and then suspended in phenol red-free DMEM/F12 at the concentrations indicated. Treatment with 0.03% DMSO equivalent to 30 μM XN had no effect on E2-induced cell growth, ERα-target gene expression, Akt (S473), MAPK (T202/Y204) and ERα (S104/S106, S118, S167, S305 and Y537) activities and BIG3-PHB2 interactions (Supplementary Fig. S5a–d). A dominant-negative peptide (ERAP; 11R-GGG-QMLSDLTLQLRQR), which was designed to specifically inhibit the BIG3-PHB2 interaction, was synthesised as described previously9 and used as a positive control. To examine the effects of XN on the inhibition of endogenous BIG3-PHB2 complex formation, MCF-7 cells were treated with 10 nM E2 ± XN. The BIG3-PHB2 interactions were assessed 24 h later using co-immunoprecipitation followed by immunoblot analysis, as described above.
Nuclear/cytoplasmic fractionation
Nuclear and cytoplasmic fractions of MCF-7 cells were prepared using the NE-PER nuclear and cytoplasmic extraction reagent (Thermo Fisher Scientific) as described previously9. α/β-Tubulin and laminin B were used as loading controls for the cytoplasmic and nuclear fractions, respectively.
Immunocytochemical staining measurement of the nuclear-translocation of PHB2
MCF-7 cells were seeded at 5 × 104 cells per well in 8-well chambers (Laboratory-Tek II Chamber Slide System, Nalgene, Nunc International) for 48 h and then treated with E2 ± XN or ERAP for 24 h. The staining procedures were conducted as described previously9.
Luciferase reporter assay
Transfections of ERE-luciferase reporter into MCF-7 or KPL-3C cells were performed by ERE reporter assay kit (Qiagen). The enclosed Renilla-luciferase reporter was monitored as an internal control. Briefly, 16 h post-transfection, the culture medium was changed to assay medium (Opti-MEM, 10% FBS, 0.1 mM NEAA, 1 mM sodium pyruvate and 10 μg mL−1 insulin). After 8 h, the cells were exposed to E2 ± XN for 24 h. Then, the cells were harvested and analysed for luciferase and Renilla-luciferase activities using the Promega dual luciferase reporter assay (Promega KK, Tokyo, Japan) as described previously9.
Cell proliferation assay
The cell proliferation assays were performed using the Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) as described previously9. The data represent the mean ± SE of three independent experiments.
Real-time PCR
The expression of the ERα target genes (TFF1 and CCND1) and endoplasmic reticulum-responsive genes (CHOP and GRP78) was evaluated by real-time RT-PCR as described previously9. Each sample was normalised to the β2-MG mRNA content and the results were expressed as the fold increase over untreated cells at 0 h (set at 1.0). The data represent the mean ± SE of three independent experiments. The primers were as follows: TFF1 5′-GGCCTCCTTAGGCAAATGTT-3′ and 5′-CCTCCTCTCTGCTCCAAAGG-3′, CCND1 5′-CAGAAGTGCGAGGAGGAGGT-3′ and 5′-CGGATGGAGTTGTCGGTGT-3′, CHOP 5′-CTGGAAGCCTGGTATGAGGA-3′ and 5′-CAGTCAGCCAAGCCAGAGA-3′, GRP78 5′-CCCCAACTGGTGAAGAGG-3′ and 5′-CCCAAAGACATGTGAGCAA-3′ and β2-MG 5′-AACTTAGAGGTGGGGAGCAG-3′ and 5′-CACAACCATGCCTTACTTTATC-3′.
Cell cycle assay
The cell cycle assay was performed as described previously9. The cells were fixed in ice cold 70% ethanol, incubated with 20 μg mL−1 propidium iodide (Sigma) and 1 mg mL−1 ribonuclease A (Sigma) and analysed by flow cytometry using a FACSCalibur with CellQuest software (BD, Franklin Lakes, NJ, USA).
In vivo tumour growth inhibition
KPL-3C cell suspensions (1 × 107 cells per mouse) were mixed with an equal volume of Matrigel (BD) and injected (200 μL total) into the mammary fat pads of 6-week-old female BALB/c nude mice (Charles River Laboratories, Tokyo, Japan). The mice were housed in a pathogen-free isolation facility with a 12-h light/dark cycle and were fed rodent chow and water ad libitum. The tumours developed over a period of a few days, reaching sizes of approximately 100 mm3 [calculated as 1/2 × (width × length2)]. The mice were randomised into five treatment groups (five animals per group): 1) no treatment; 2) 6 µg per day E2; 3) E2 + 0.3 mg kg−1 per day XN; 4) E2 + 1.0 mg kg−1 per day XN; 5) E2 + 14 mg kg−1 per day ERAP. E2 was delivered via the application of a solution to the neck skin; the other treatments were delivered via intraperitoneal injection. The tumour volume was measured with calipers for 36 days, after which time the animals were sacrificed and the tumours were excised. All of the experiments were performed in accordance with the guidelines of the animal facility at the University of Tokushima.
Immunohistochemical staining of xenografts
To examine the PHB2, Ki67 and PCNA protein expression in KPL-3C xenograft tumours, we stained 3-μm sections of paraffin-embedded tumours with anti-PHB2 (dilution 1:250), anti-Ki67 (dilution 1:50) or anti-PCNA (diluted 1:200) antibodies, as previously described39. The nuclear intensities of the PHB2, Ki67 and PCNA staining were determined using MetaMorph software (Molecular Devices, Tokyo, Japan); the data represent the mean ± SE of five different points.
Statistical analyses
Student's t-test was used to determine the significance of the differences among the experimental groups. Values of P < 0.05 were considered significant.
References
Johnston, S. R. New strategies in estrogen receptor-positive breast cancer. Clin. Cancer Res. 16, 1979–1987 (2010).
Fisher, B. et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer Inst. 97, 1652–1662 (2005).
Jordan, V. C. Tamoxifen: a most unlikely pioneering medicine. Nature Rev. Drug Discov. 2, 205–213 (2003).
Clarke, R., Leonessa, F., Welch, J. N. & Skaar, T. C. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol. Rev. 53, 25–71 (2001).
Fisher, B., Dignam, J., Bryant, J. & Wolmark, N. Five versus more than five years of tamoxifen for lymph node-negative breast cancer: updated findings from the National Surgical Adjuvant Breast and Bowel Project B-14 randomized trial. J. Natl. Cancer Inst. 93, 684–690 (2001).
Chlebowski, R. et al. Clinical perspectives on the utility of aromatase inhibitors for the adjuvant treatment of breast cancer. Breast 2, S1–11 (2009).
Chumsri, S., Howes, T., Bao, T., Sabnis, G. & Brodie, A. Aromatase, aromatase inhibitors and breast cancer. J. Steroid Biochem. Mol. Biol. 125, 13–22 (2011).
Kim, J. W. et al. Activation of an estrogen/estrogen receptor signaling by BIG3 through its inhibitory effect on nuclear transport of PHB2/REA in breast cancer. Cancer Sci. 100, 1468–1478 (2009).
Yoshimaru, T. et al. Targeting BIG3-PHB2 interaction to overcome tamoxifen resistance in breast cancer cells. Nat. Commun. 4, 2443 (2013).
Pan, L., Becker, H. & Gerhauser, C. Xanthohumol induces apoptosis in cultured 40-16 human colon cancer cells by activation of the death receptor- and mitochondrial pathway. Mol. Nutr. Food Res. 49, 837–843 (2005).
Deeb, D. et al. Growth inhibitory and apoptosis-inducing effects of xanthohumal, a prenylated chalone present in hops, in human posthaste cancer cells. Anticancer Res. 30, 3333–3339 (2010).
Gerhauser, C. Beer constituents as potential cancer chemopreventive agents. Eur. J. Cancer 41, 1941–1954 (2005).
Vanhoecke, B. et al. Antiinvasive effect of xanthohumol, a prenylated chalcone present in hops (Humulus lupulus L.) and beer. Int. J. Cancer 117, 889–895 (2005).
Zanoli, P. & Zavatti, M. Pharmacognostic and pharmacological profile of Humulus lupulus L. J. Ethnopharmacol. 116, 383–896 (2008).
Blanquer-Rosselló, M. M., Oliver, J., Valle, A. & Roca, P. Effect of xanthohumol and 8-prenylnaringenin on MCF-7 breast cancer cells oxidative stress and mitochondrial complexes expression. J. Cell Biochem. 114, 2785–2794 (2013).
Rosário, M., Ana, F., Isabel, A. & Conceição, C. Modulation of breast cancer cell survival by aromatase inhibiting hop (Humulus lupulus L.) flavonoids. J. Steroid Biochem. Mol. Biol. 124, 124–130 (2007).
Sasazawa, Y. et al. Xanthohumol impairs autophagosome maturation through direct inhibition of valosin-containing protein. ACS Chem. Biol. 7, 892–900 (2012).
Kahlert, S. et al. Estrogen receptor rapidly activates the IGF-1 receptor pathway. J. Biol. Chem. 275, 18447–18453 (2000).
Simoncini, T. et al. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407, 538–541 (2000).
Castoria, G. et al. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 20, 6050–6059 (2001).
Song, R. X. et al. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor α to the plasma membrane. Proc. Natl. Acad. Sci. USA 101, 2076–2081 (2004).
Murphy, L. C., Seekallu, S. V. & Watson, P. H. Clinical significance of estrogen receptor phosphorylation. Endocrine-Related Cancer 18, R1–R14 (2011).
Lannigan, D. A. Estrogen receptor phosphorylation. Steroids 68, 1–9 (2008).
Chen, D. et al. Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol. Cell 6, 127–137 (2000).
Chen, D., Pace, P. E., Coombes, R. C. & Ali, S. Phosphorylation of human estrogen receptor alpha by protein kinase A regulates dimerization. Mol. Cell Biol. 19, 1002–1015 (1999).
Joel, P. B. et al. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol. Cell Biol. 18, 1978–1984 (1998).
Rogatsky, I., Trowbridge, J. M. & Garabedian, M. J. Potentiation of human estrogen receptor alpha transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin A-CDK2 complex. J. Biol. Chem. 274, 22296–22302 (1999).
Arnold, S. F., Melamed, M., Vorojeikina, D. P., Notides, A. C. & Sasson, S. Estradiol-binding mechanism and binding capacity of the human estrogen receptor is regulated by tyrosine phosphorylation. Mol. Endocrinol. 11, 48–53 (1997).
Wang, R. A., Mazumdar, A., Vadlamudi, R. K. & Kumar, R. P21-activated kinase-1 phosphorylates and transactivates estrogen receptor-alpha and promotes hyperplasia in mammary epithelium. EMBO J. 21, 5437–5447 (2001).
Strathmann, J. et al. Xanthohumol from hops prevents hormone-dependent tumorigenesis in vitro and in vivo. Acta Hort. 848, 179–190 (2009).
Unek, G., Ozmen, A., Ozekinci, M., Sakinci, M. & Korgun, E. T. Immunolocalization of cell cycle proteins (p57, p27, cyclin D3, PCNA and Ki67) in intrauterine growth retardation (IUGR) and normal human term placentas. Acta Histochem. 116, 493–502 (2014).
Kurki, P., Lotz, M., Ogata, K. & Tan, E. M. Proliferating cell nuclear antigen (PCNA)/cyclin in activated human T lymphocytes. J. Immunol. 138, 4114–4120 (1987).
Gerhauser, C. et al. Cancer chemopreventive activity of Xanthohumol, a natural product derived from hop. Mol. Cancer Ther. 1, 959–969 (2002).
Gao, X. et al. Immunomodulatory activity of xanthohumol: inhibition of T cell proliferation, cell-mediated cytotoxicity and Th1 cytokine production through suppression of NF-κB. Immunophamacol. Immunotoxicol. 31, 477–484 (2009).
Benelli, R. et al. The AKT/NF-κB inhibitor xanthohumol is a potent anti-lymphocytic leukemia drug overcoming chemoresistance and cell infiltration. Biochem. Pharmacol. 83, 1634–1642 (2012).
Yamamoto, S. et al. Expression level of valosin-containing protein is strongly associated with progression and prognosis of gastric carcinoma. J. Clin. Oncol. 21, 2537–2544 (2003).
Yamamoto, S. et al. Increased expression of valosin-containing protein (p97) is correlated with disease recurrence in follicular thyroid cancer. Ann. Surg. Oncol. 12, 925–934 (2005).
Kurebayashi, J., Kurosumi, M. & Sonoo, H. A new human breast cancer cell line, KPL-3C, secretes parathyroid hormone-related protein and produces tumours associated with microcalcifications in nude mice. Br. J. Cancer 74, 200–207 (1996).
Shan, H., Takahashi, T., Bando, Y., Izumi, K. & Uehara, H. Inhibitory effect of soluble platelet-derived growth factor receptor β on intraosseous growth of breast cancer cells in nude mice. Cancer Sci. 102, 1904–1910 (2011).
Acknowledgements
The authors thank Dr. Jun-ichi Kurebayashi (Kawasaki Medical School) for the gifts of the KPL-3C breast cancer cell line. This work was supported by the Project Future of Relay For Life Japan, a grant/research support from Tokushima Breast Care Clinic, a Grant-in-Aid for Scientific Research on Innovative Areas (MEXT KAKENHI Grant Number 251347212), Grants-in-Aid for Scientific Research (B) (MEXT KAKENHI Grant Number 25293079) and (C) (MEXT KAKENHI Grant Number 26461948) and IMSUT Joint Research Project.
Author information
Authors and Affiliations
Contributions
T.Y. performed all of the experiments and prepared the draft of the manuscript. M.K. performed the in vivo experiments. E.T., M.I. and H.O. performed isolation and purification of XN and in vitro binding assay for the direct binding of XN to recombinant PHB2 protein. Y.M., J.H. and M.S. discussed the interpretation of antitumor effect of XN or ERAP on the ERα-signalling pathway in breast cancer. T.K. was involved in the conception and design of all of the studies, the interpretation of data and preparing the draft and final version of the manuscript. All of the authors read and approved the final manuscript.
Ethics declarations
Competing interests
Toyomasa Katagiri is a stock holder and an external board member of OncoTherapy Science, Inc. The other authors have declared that no conflicts of interest exist.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yoshimaru, T., Komatsu, M., Tashiro, E. et al. Xanthohumol suppresses oestrogen-signalling in breast cancer through the inhibition of BIG3-PHB2 interactions. Sci Rep 4, 7355 (2014). https://doi.org/10.1038/srep07355
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07355
This article is cited by
-
Semi-synthesis and in vitro anti-cancer effects evaluation of novel xanthohumol derivatives
Molecular Diversity (2023)
-
Anticancer properties of medicinal plants and their bioactive compounds against breast cancer: a review on recent investigations
Environmental Science and Pollution Research (2022)
-
Prohibitin ligands: a growing armamentarium to tackle cancers, osteoporosis, inflammatory, cardiac and neurological diseases
Cellular and Molecular Life Sciences (2020)
-
Gut Microbiota as a Positive Potential Therapeutic Factor in Carcinogenesis: an Overview of Microbiota-Targeted Therapy
Journal of Gastrointestinal Cancer (2020)
-
Significance of prohibitin domain family in tumorigenesis and its implication in cancer diagnosis and treatment
Cell Death & Disease (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
