
Abstract
We report herein the synthesis and application of a series of novel cyclometalated iridium(III) complexes 1−3 bearing a rhodamine-linked NˆN ligand for the detection of Cu2+ ions. Under the optimised conditions, the complexes exhibited high sensitivity and selectivity for Cu2+ ions over a panel of other metal ions and showed consistent performance in a pH value range of 6 to 8. Furthermore, the potential application of this system for the monitoring of Cu2+ ions in tap water or natural river water samples was demonstrated.
Similar content being viewed by others
Introduction
The Cu2+ ion plays an important role in a number of biological processes, including iron absorption, haemopoiesis, various enzyme-catalyzed and redox reactions1. Abnormal levels of copper ions can lead to vomiting, lethargy, increased blood pressure and respiratory rates, acute haemolytic anaemia, liver damage, neurotoxicity and neurodegenerative disease2,3. Furthermore, copper ions can disrupt natural ecosystems due to their adverse effects on microorganisms4. Therefore, the development of selective and reliable detection methods for Cu2+ ions is of significant interest to the scientific and environmental communities.
A number of analytical methods have been developed for the accurate determination of Cu2+ ions, including atomic absorption/emission spectroscopy (AAS/AES)5, inductively-coupled plasma mass spectrometry (ICP-MS)6, electrochemical methods7 and X-ray fluorescence spectroscopy (XRF). However, these techniques involve time-consuming sample pre-treatment methods and/or the use of sophisticated instrumentation, thus precluded their application for in-field detection of Cu2+ ions. This has stimulated the development of a variety of small molecule probes for the rapid and sensitive detection of Cu2+ ions4,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22 (Table 1).
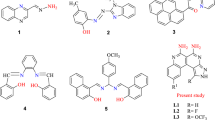
Transition metal complexes have found increasing use as colorimetric or luminescent probes for biomolecules and metal ions23,24,25,26,27,28,29,30,31,32,33,34,35. In particular, octahedral metal complexes possess a unique geometry around which functional co-ligands can be arranged in a defined fashion. For example, Li, Huang and co-workers have previously developed a dual colorimetric and luminescent iridium(III)-based sensor for Hg2+ ions by the use of sulfur-containing cyclometalated ligands24. In the context of Cu2+ ion detection, several groups have reported DPA-based iridium(III) complexes for the luminescent sensing of Cu2+ ions25,36. However, to our knowledge, no colorimetric iridium(III)-based Cu2+ ion probe has yet been described in the literature. We report herein the synthesis and application of a series of novel cyclometalated iridium(III) complexes 1–3 bearing a rhodamine-linked NˆN ligand for the highly sensitive and selective detection of Cu2+ ions (Figure 1).

Chemical structures of cyclometallated iridium(III) complexes 1–3.
The synthetic pathway for the synthesis of complexes 1–3 is depicted in Scheme S1. In our design strategy, the interaction of Cu2+ ions with the rhodamine-linked NˆN ligand causes the rhodamine moiety to undergo a conformational change from a spirolactam form into a ring-opened amide form9. This structural transition results in a change to the photophysical properties of these complexes, allowing them to function as optical chemosensors for Cu2+ ions.
Results
Cu2+ ion detection
To investigate the Cu2+ ion detection properties of complexes 1–3, we studied the UV-Vis absorption behaviour of these complexes in response to Cu2+ ions. Remarkably, a new absorption peak at 555 nm was observed in the spectra of complexes 1–3 (5 μM) upon the addition of Cu2+ ions (Figure S1) and the color of the solution turned pink. This behavior was attributed to the interaction of Cu2+ ions with the rhodamine-linked NˆN ligand, causing the rhodamine moiety to undergo a conformational change from a spirolactam form into a ring-opened amide form (Figure 1), giving rise to a pink color that is consistent with previous work9. Moreover, the UV-Vis spectra showed that the complexes were stable in the absence or presence of Cu2+ ions for at least 24 h (Figure S2).
To optimize the performance of the sensor, the choice of organic solvent, aqueous buffer and overall solvent composition were investigated. The absorption increase of complexes 1–3 in response to Cu2+ ions was highest with acetonitrile (ACN) compared to dimethyl sulfoxide (DMSO), tetrahydrofuran (THF) or N,N-dimethylformamide (DMF) (Figure S3). Additionally, complexes 1–3 displayed similar responses to Cu2+ ions in buffer systems containing 90, 70 or 50% of acetonitrile (Figure S4). However, the absorption of the complexes was seen to decrease by 30-50% when 30% of acetonitrile was used (Figure S4). Hence, 50% of acetonitrile was considered optimal for further study. We also found that the use of 2-amino-2-hydroxymethyl-propane-1,3-diol (Tris), 2-(N-morpholino)ethanesulfonic acid (MES) and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer gave similar performances, whereas the use of phosphate buffer resulted in a 25% decrease in absorption intensity of the complexes in response to Cu2+ ions (Figure S5). Moreover, the optimal concentration of Tris buffer was determined to be 10 mM (Figure S6). After optimization of the assay conditions, we performed an absorption titration experiment to investigate the absorption response of complexes 1–3 to Cu2+ ions. Encouragingly, the absorption of complexes 1–3 was enhanced as the concentration of Cu2+ ions increased (Figure 2a, c, e). Saturation of the absorbance value was reached at 35 μM of Cu2+ ions for all three complexes. Complexes 1 and 3 exhibited a linear range of detection for Cu2+ ions from 10 nM to 8 μM (R2 = 0.99), while complex 2 displayed a linear range of detection for Cu2+ ions from 10 nM to 6 μM (Figure 2b, d, f). The detection limit of complexes 1–3 for Cu2+ ions was estimated to be 4.5, 5.2 and 4.9 nM using the 3σ criterion, indicating that these complexes were highly sensitive for Cu2+ ions. Additionally, the color change of the solution from colorless to pink occurred within 10 s upon the addition of Cu2+ ions, suggesting that complexes 1–3 could potentially serve as simple and rapid ‘naked-eye’ indicators for Cu2+ ions (Figure 3a).

Absorption spectra of 5 μM of complexes (a) 1, (c) 2 and (e) 3 in the presence of increasing concentrations of Cu2+ ion (1:1 ACN-Tris, pH 7.0). Absorbance of complexes (b) 1, (d) 2 and (f) 3 at 555 nm vs. [Cu2+]. Inset: linear plot of the change in absorbance of the system vs. [Cu2+].

Photograph images of complex 1 (5 μM) in the presence of (a) 0, 1, 3, 5, 10 and 20 μM of Cu2+ (left to right) and (b) 25 μM of Zn2+, Cd2+, Mg2+, Ag+, Hg2+, Ca2+, Pb2+, Ni2+, Co2+, Fe3+, K+ and Na+ or 10 μM of Cu2+ (left to right).
Selectivity analysis
The selectivity of complexes 1–3 for Cu2+ ions was examined by testing the response of the system to various other metal ions, including Zn2+, Cd2+, Mg2+, Hg2+, Ca2+, Pb2+, Ni2+, Co2+, Fe3+, K+, Na+ and Ag+. In the absence of metal ions, the UV-Vis absorption spectra of complexes 1–3 displayed no significant absorption in the 400 to 600 nm region (Figure 4). The addition of 10 μM of Cu2+ ions to complexes 1–3 generated a new absorption band at 555 nm, with a 50-fold increase in absorbance signal. By comparison, only minor effects on the absorbance signals of complex 1–3 were observed upon the addition of 25 μM of the other metal ions. Importantly, the difference in absorbance intensity of complex 1 in the presence of Cu2+ ions compared to the other metal ions could be distinguished by the naked eye (Figure 3b). These results demonstrate the high selectivity of complexes 1–3 for Cu2+ ions over other metal ions, which originates presumably from the specific interaction of Cu2+ ions with the rhodamine-linked NˆN ligand. The rhodamine moiety is known to be highly selective for Cu2+ ions over other common metal ions9. A competition experiment was also carried out by adding 25 μM of other metal ions to solutions of complexes 1–3 containing 10 μM of Cu2+ ions. The absorbance intensity of complexes 1–3 was not significantly abrogated by the presence of the other metal ions (Figure 5), suggesting that the system could potentially be used to detect Cu2+ ions in a sample matrix containing interfering metal ions.

UV-Vis spectra of 5 μM of complexes 1 (a), 2 (c) and 3 (e) in the presence of 10 μM of Cu2+ or 25 μM of various metal ions (1:1 ACN-Tris, pH 7.0). Absorbance of 5 μM of complexes 1 (b), 2 (d) and 3 (f) at 555 nm in the presence of 10 μM of Cu2+ and 25 μM of various metal ions.

Absorbance of 5 μM of complexes (a) 1, (b) 2 and (c) 3 at 555 nm in the presence of both 10 μM of Cu2+ and 25 μM of various metal ions (1:1 ACN-Tris, pH 7.0).
Job's plot analysis
The absorbance spectra were analysed using a Job's plot to determine the binding stoichiometry of the iridium(III) complexes 1–3 with Cu2+ ions. The maximum absorbance of the 1–Cu2+, 2–Cu2+ and 3–Cu2+ complexes were all achieved at a mole fraction of approximately 50% of Cu2+ ions (Figure 6), suggesting that a 1:1 ratio is the likely binding stoichiometry of complexes 1–3 with Cu2+ ions.

Job's plot of complexes (a) 1, (b) 2 and (c) 3 with Cu2+ ions (1:1 ACN-Tris, pH 7.0).
Total concentration of 5 μM, λabs = 555 nm.
Influence of pH
We next investigated the effect of pH on the response of complexes 1–3 to Cu2+ ions. The results showed that the performances of the complexes were quite consistent over a pH range of 6 to 8 (Figure S7). This result is consistent with a previous study by Yin and co-workers, who showed that a rhodamine-based sensor could function normally under this pH range22. Hence, complexes 1–3 are suitable for use as Cu2+ chemosensors in the typical pH range of biological systems.
Cu2+ ion detection in a tap water sample
In order to investigate the applicability of the system for real sample analysis, we investigated the ability of complexes 1–3 to detect Cu2+ ions in three water samples using the spike-and-recovery method. Two of the samples were tap water taken from laboratory water pipe located at Hong Kong Baptist University, Hong Kong, while the third sample was a natural water sample taken from Shing Mun River, Hong Kong. The Cu2+ ion content in the water sample was first determined by atomic absorption spectroscopy. Then, 0.05 and 2 μM of Cu2+ ions was spiked into the sample. Using iridium(III) complexes 1–3 as Cu2+ ion probes, the recovery factor was determined to be ca. 80−110% for Cu2+ ion concentrations in the middle region of the linear range of detection and ca. 60−130% for Cu2+ ion concentrations close to the detection limit (Table 2)37. This result demonstrates the potential application of the system in monitoring Cu2+ ion content in real water samples. Although the detection limit of complexes 1–3 for Cu2+ ions in the buffer/organic solvent mixture system is much lower than those reported in other studies, the accuracy of the complexes in real samples seems worse. This may be attributed to the influence of interfering matrix substances in the real water samples, which is a significant factor especially when the spiked Cu2+ concentration in this study is as low as 50 nM, compared to 3, 30 or 160 μM that was used in other studies (Table 1).
Discussion
In summary, we have developed a series of novel iridium(III) complexes 1–3 as colorimetric probes for the sensitive detection of Cu2+ ions and have demonstrated the potential application of the system for the monitoring of Cu2+ ions in real life water samples. Under the optimised conditions, the complexes exhibited high selectivity for Cu2+ ions over a panel of other metal ions and showed consistent performance over a pH range of 6 to 8. Our method is simple, rapid, cost-effective and could find potential application as a ‘naked-eye’ indicator for Cu2+ ions in water samples. Due to the modular nature of metal complex synthesis, we envisage that this iridium-based chemosensor for Cu2+ ions could be further readily fine-tuned without the need for labor-intensive synthetic protocols. We anticipate that this report of a colorimetric iridium(III)-based chemosensor for Cu2+ ions could prompt the development of more sensitive and cost-effective methods for Cu2+ ion detection from the scientific community.
Methods
Chemicals and materials
Reagents were purchased from Sigma Aldrich (St. Louis, MO) and were used as received. Iridium chloride hydrate (IrCl3.xH2O) was purchased from Precious Metals Online (Australia).
General experimental
Mass spectrometric measurements were performed at the Mass Spectroscopy Unit at the Department of Chemistry, Hong Kong Baptist University, Hong Kong (China). Melting points were determined using a Gallenkamp melting apparatus and are uncorrected. Deuterated solvents for NMR purposes were obtained from Armar and used as received. 1H and 13C NMR were recorded on a Bruker Avance 400 spectrometer operating at 400 MHz (1H) and 100 MHz (13C). 1H and 13C chemical shifts were referenced internally to solvent shift (CD3CN: 1H, δ 1.94, 13C δ 118.7; d6-DMSO: 1H δ 2.50, 13C δ 39.5). Chemical shifts (δ) are quoted in ppm, the downfield direction being defined as positive. Uncertainties in chemical shifts are typically ±0.01 ppm for 1H and ±0.05 for 13C. Coupling constants are typically ±0.1 Hz for 1H-1H and ±0.5 Hz for 1H-13C couplings. The following abbreviations are used for convenience in reporting the multiplicity of NMR resonances: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. All NMR data was acquired and processed using standard Bruker software (Topspin).
Cu2+ ion detection
10 mM of complex stock solution was prepared by dissolving complexes 1–3 in acetonitrile. The complexes were then added into 1:1 (v/v) acetonitrile: Tris buffer to a final concentration of 5 μM. Different concentrations of Cu2+ ions (as Cu(NO3)2) were then added to 1 mL of 1:1 (v/v) acetonitrile: Tris buffer (10 mM, pH 7.0) containing complexes 1–3 (5 μM) in a cuvette. UV-Vis absorption spectra were recorded on a Cary UV-300 spectrophotometer (double beam).
Water sample analysis
Two tap water samples were taken from a laboratory water pipe located in Hong Kong Baptist University, Hong Kong and a river water sample was taken from Shing Mun River, Hong Kong. Standard solution of Cu2+ ions was spiked into the real water sample up to a final concentration of 2 μM of Cu2+ ions. 0.5 mL of the resulting solution was mixed with 0.5 mL of acetonitrile in a cuvette and then complexes 1–3 were added at a final concentration of 5 μM. UV-Vis absorption spectra were recorded on a Cary UV-300 spectrophotometer (double beam).
Synthesis of compounds 5 and 6
These compounds were synthesised according to a reported literature procedure38. Following a Steck-Day reaction procedure, 500 mg of 1,10-phenanthroline-5,6-dione (2.4 mmol) and 350 mg of 4-hydroxybenzaldehyde (2.8 mmol, 1.1 eq.) were dissolved in 10 mL of acetic acid. The mixture was heated at 80°C for 30 min. 3.70 g of dry ammonium acetate (48 mmol, 20 eq.) was added and the reaction was stirred at 100°C for 3 h. The reaction was allowed to cool to room temperature and the product was filtered through a fritted funnel and washed with copious amounts of acetone and ether to yield 510 mg (68%) of compound 5. Following a modified Duff reaction, 500 mg (1.6 mmol) of 5 and 896 mg of hexamethylene tetraamine (6.4 mmol, 4 eq.) were dissolved in 10 mL of trifluoroacetic acid and refluxed for 3 days. The mixture was allowed to cool to room temperature and 50 mL of 4 M HCl solution was added. The resulting mixture was stirred for 2 h, during which time the solution became cloudy. The solid was collected by filtration, washed with acetone and ether and dried in vacuum to yield 452 mg (56%) of pure compound 6.
Synthesis of compound 8
This compound was synthesised according to a reported literature procedure9. Rhodamine B (2.5 mmol) was dissolved in 30 mL of ethanol and 3.0 mL of hydrazine hydrate (85%, excess) was added dropwise with vigorous stirring at room temperature. The mixture was heated to reflux in air for 2 h with stirring. The solution changed from dark purple to light orange and then became clear. The mixture was cooled and the solvent was removed under reduced pressure. 1 M HCl (50 mL) was added to the solid in the flask to generate a clear red solution. The solution was then basified by the slow addition of 1 M NaOH until the pH of the solution reached 9–10. The resulting precipitate was filtered and washed three times with 15 mL of water. Drying under an IR light afforded 0.83 g (75%) of 8 as a pink solid.
Synthesis of compound 9
This compound was synthesised according to a reported literature procedure9. Rhodamine hydrazide (8, 1 mmol) was dissolved in 30 mL of absolute ethanol. An excess amount of compound 6 (4 mmol) was added and the mixture was refluxed in air for 6 h. The solution was cooled, concentrated to 10 mL and allowed to stand at room temperature overnight. The precipitate which appeared the next day was filtered and washed three times with 10 mL of cold ethanol. Drying under reduced pressure afforded 0.58 g (74%) of 9 as a pink solid. Yield: 65%. 1H NMR (400 MHz, CDCl3) δ 13.15 (s, 1H), 11.32 (s, 1H), 9.00 (s, 1H), 8.93 (d, J = 7.7 Hz, 2H), 8.84 (d, J = 3.2 Hz, 1H), 8.62 (d, J = 7.8 Hz, 1H), 7.89 (t, J = 7.7 Hz, 2H), 7.82 (s, 1H), 7.59–7.47 (m, 2H), 7.43 (t, J = 7.3 Hz, 1H), 7.31 (dd, J = 7.8, 4.2 Hz, 1H), 7.15 (d, J = 7.6 Hz, 1H), 6.69 (d, J = 8.6 Hz, 1H), 6.52 (d, J = 8.9 Hz, 2H), 6.46 (d, J = 2.4 Hz, 2H), 6.24 (dd, J = 9.0, 2.4 Hz, 2H), 3.25 (d, J = 7.4 Hz, 8H), 1.09 (t, J = 7.0 Hz, 12H); 13C NMR (100 MHz, CDCl3) δ 164.49, 159.52, 153.49, 152.31, 151.00, 149.10, 147.59, 143.53, 136.00, 133.74, 130.44, 129.96, 129.17, 128.65, 126.53, 124.10, 123.38, 122.74, 121.49, 119.66, 118.64, 117.43, 108.13, 104.95, 98.06, 66.65, 58.15, 44.30, 30.97, 18.36; HRMS: Calcd. for C48H42N8O3: 778.3380, Found: 779.4721.
Synthesis of complexes 1–3
The precursor complexes [Ir2(ppy)4Cl2], [Ir2(dfppy)4Cl2] and [Ir2(bzq)4Cl2] and the metal complexes 1–3 were prepared according to modified literature methods39.
Representative procedure for complex 1: A suspension of [Ir2(ppy)4Cl2] (0.2 mmol) and compound 6 (0.44 mmol) in 20 mL dimethylformamide was refluxed overnight under a nitrogen atmosphere. The resulting solution was allowed to cool to room temperature. An aqueous solution of ammonium hexafluorophosphate (excess) was added to the mixture and the resulting precipitate was then filtered. The crude product was purified by silica gel chromatography with a dichloromethane-methanol solvent system (30:1 v/v) as eluent. The product was obtained as a brown-orange solid.
Complex 1. Yield: 55%. 1H NMR (400 MHz, Acetone-d6) δ 9.46 (s, 1H), 9.06 (d, J = 4.0 Hz, 2H), 8.34 (d, J = 4.0 Hz, 2H), 8.27 (d, J = 8.0 Hz, 2H), 8.12 (t, J = 8.0 Hz, 2H), 7.96–7.89 (m, 7H), 7.70–7.74 (m, 3H), 7.67 (t, J = 8.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.10 (t, J = 8.0 Hz, 2H), 7.02–6.96 (m, 5H), 6.59–6.42 (m, 8H), 3.43–3.37 (m, 8H), 1.17 (t, J = 8.0 Hz, 12H); 13C NMR (100 MHz, Acetone-d6) δ 168.7, 165.0, 161.1, 160.8, 154.5, 153.3, 152.6, 151.9, 151.3, 150.3, 150.1, 149.7, 145.1, 139.5, 134.9, 133.0, 132.7, 131.3, 130.5, 130.4, 130.2, 129.9, 129.0, 128.9, 127.5, 127.4, 125.8, 125.1, 124.4, 124.1, 123.4, 121.8, 120.8, 119.8, 118.5, 109.3, 106.1, 98.6, 67.3, 44.8, 12.7 HRMS: Calcd. for C70H58IrN10O3 [M–PF6]+:1279.4917 Found: 1278.7978.
Complex 2. Yield: 57%. 1H NMR (400 MHz, Acetone-d6) δ 9.49 (s, 1H), 9.20 (d, J = 8.0 Hz, 2H), 8.48 (d, J = 4.0 Hz, 4H), 8.14–7.96 (m, 7H), 7.81 (s, J = 4.0 Hz, 2H), 7.73 (t, J = 8.0 Hz, 2H), 7.67 (t, J = 8.0 Hz, 2H), 7.22 (s, J = 8.0 Hz, 1H), 7.08 (t, J = 8.0 Hz, 2H), 6.85 (t, J = 10.0 Hz, 2H), 6.60–6.52 (m, 4H), 6.45–6.42 (m, 2H), 5.92 (d, J = 8.0 Hz, 2H), 3.42 (d, J = 8.0 Hz, 8H), 1.18–1.13 (m, 12H); 13C NMR (100 MHz, Acetone-d6) δ 164.2, 163.9, 163.8, 162.7, 162.6, 160.0, 154.6, 153.5, 152.8, 150.0, 149.2, 144.2, 139.7, 134.1, 132.7, 129.7, 129.2, 128.9, 128.0, 127.9, 126.9, 124.2, 124.1, 123.7, 123.5, 123.4, 123.1, 121.1, 117.8, 117.5, 114.0, 113.8, 108.4, 99.1, 98.8, 98.5, 97.7, 66.4, 45.8, 44.0, 11.9; HRMS: Calcd. for C70H54F4IrN10O3 [M–PF6]+: 1351.4536, Found: 1350.8763.
Complex 3. Yield: 58%. 1H NMR (400 MHz, CDCl3) δ 14.23 (s, 1H), 10.87 (s, 1H), 9.08 (s, 2H), 8.95 (s, 1H), 8.54 (d, J = 7.7 Hz, 2H), 8.35 (s, 1H), 8.12 (s, 2H), 8.08–7.83 (m, 10H), 7.62 (dd, J = 19.6, 7.6 Hz, 4H), 7.47 (s, 2H), 7.23 (t, J = 7.0 Hz, 2H), 7.09 (dd, J = 26.3, 8.0 Hz, 2H), 6.60–6.24 (m, 8H), 3.32 (s, 8H), 1.07 (t, J = 6.4 Hz, 12H); 13C NMR (100 MHz, CDCl3) δ 163.8, 162.3, 158.8, 156.3, 152.6, 152.3, 151.5, 148.9, 148.6, 147.1, 144.4, 140.3, 137.5, 136.6, 134.2, 133.7, 131.8, 129.7, 129.4, 128.9, 128.5, 127.9, 127.6, 126.7, 124.2, 123.8, 123.2, 122.7, 120.9, 120.3, 119.9, 117.1, 108.2, 108.1, 104.5, 97.4, 65.2, 54.9, 45.4, 43.6, 35.7, 30.7, 12.4; HRMS: Calcd. for C74H58IrN10O3 [M–PF6]+: 1327.5345 Found: 1326.9231.
References
Malvankar, P. L. & Shinde, V. M. Ion-pair extraction and determination of copper(II) and zinc(II) in environmental and pharmaceutical samples. Analyst 116, 1081–1084 (1991).
Vulpe, C., Levinson, B., Whitney, S., Packman, S. & Gitschier, J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 3, 7–13 (1993).
Bull, P. C., Thomas, G. R., Rommens, J. M., Forbes, J. R. & Cox, D. W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 5, 327–337 (1993).
Krämer, R. Fluorescent Chemosensors for Cu2+ Ions: Fast, Selective and Highly Sensitive. Angew. Chem. Int. Ed. 37, 772–773 (1998).
Tong, A., Akama, Y. & Tanaka, S. Pre-concentration of copper, cobalt and nickel with 3-methyl-1-phenyl-4-stearoyl-5-pyrazolone loaded on silica gel. Analyst 115, 947–949 (1990).
Rao, G. P. C., Seshaiah, K., Rao, Y. K. & Wang, M. C. Solid Phase Extraction of Cd, Cu and Ni from Leafy Vegetables and Plant Leaves Using Amberlite XAD-2 Functionalized with 2-Hydroxy-acetophenone-thiosemicarbazone (HAPTSC) and Determination by Inductively Coupled Plasma Atomic Emission Spectroscopy. J. Arg. Food. Chem. 54, 2868–2872 (2006).
Poursaberi, T. et al. The Synthesis of a New Thiophene-Derivative Schiff's Base and Its Use in Preparation of Copper-Ion Selective Electrodes. Electroanal. 13, 1513–1517 (2001).
Mei, L., Xiang, Y., Li, N. & Tong, A. A new fluorescent probe of rhodamine B derivative for the detection of copper ion. Talanta 72, 1717–1722 (2007).
Xiang, Y., Tong, A., Jin, P. & Ju, Y. New Fluorescent Rhodamine Hydrazone Chemosensor for Cu(II) with High Selectivity and Sensitivity. Org. Lett. 8, 2863–2866 (2006).
Xiang, Y., Li, Z., Chen, X. & Tong, A. Highly sensitive and selective optical chemosensor for determination of Cu2+ in aqueous solution. Talanta 74, 1148–1153 (2008).
Dujols, V., Ford, F. & Czarnik, A. W. A Long-Wavelength Fluorescent Chemodosimeter Selective for Cu(II) Ion in Water. J. Am. Chem. Soc. 119, 7386–7387 (1997).
Royzen, M., Dai, Z. & Canary, J. W. Ratiometric Displacement Approach to Cu(II) Sensing by Fluorescence. J. Am. Chem. Soc. 127, 1612–1613 (2005).
Zhang, X., Shiraishi, Y. & Hirai, T. Cu(II)-Selective Green Fluorescence of a Rhodamine−Diacetic Acid Conjugate. Org. Lett. 9, 5039–5042 (2007).
Yu, M. et al. Highly Sensitive and Fast Responsive Fluorescence Turn-On Chemodosimeter for Cu2+ and Its Application in Live Cell Imaging. Chem. Eur. J 14, 6892–6900 (2008).
Wu, G., Wang, D., Wu, D., Gao, Y. & Wang, Z. Highly sensitive optical chemosensor for the detection of Cu2+ using a rhodamine B spirolatam. J. Chem. Sci. 121, 543–548 (2009).
Kim, Y.-R., Kim, H. J., Kim, J. S. & Kim, H. Rhodamine-Based “Turn-On” Fluorescent Chemodosimeter for Cu(II) on Ultrathin Platinum Films as Molecular Switches. Adv. Mater. 20, 4428–4432 (2008).
Zhao, M., Yang, X.-F., He, S. & Wang, L. A rhodamine-based chromogenic and fluorescent chemosensor for copper ion in aqueous media. Sensor Actuat. B-Chem 135, 625–631 (2009).
Zhou, Y., Wang, F., Kim, Y., Kim, S.-J. & Yoon, J. Cu2+-Selective Ratiometric and “Off-On” Sensor Based on the Rhodamine Derivative Bearing Pyrene Group. Org. Lett. 11, 4442–4445 (2009).
Chen, X. et al. New fluorescent and colorimetric chemosensors bearing rhodamine and binaphthyl groups for the detection of Cu2+. Sensor Actuat B-Chem 137, 597–602 (2009).
Multhaup, G. et al. The Amyloid Precursor Protein of Alzheimer's Disease in the Reduction of Copper(II) to Copper(I). Science 271, 1406–1409 (1996).
Swamy, K. M. K. et al. Boronic acid-linked fluorescent and colorimetric probes for copper ions. Chem. Commun., 5915–5917 (2008).
Huo, F.-J. et al. A rhodamine-based dual chemosensor for the visual detection of copper and the ratiometric fluorescent detection of vanadium. Dyes Pigments 86, 50–55 (2010).
Zhao, Q., Huang, C. & Li, F. Phosphorescent heavy-metal complexes for bioimaging. Chem. Soc. Rev. 40, 2508–2524 (2011).
Zhao, Q. et al. A Highly Selective and Multisignaling Optical−Electrochemical Sensor for Hg2+ Based on a Phosphorescent Iridium(III) Complex. Organometallics 26, 2077–2081 (2007).
You, Y. et al. Phosphorescent Sensor for Robust Quantification of Copper(II) Ion. J. Am. Chem. Soc. 133, 11488–11491 (2011).
Chen, H. et al. Selective Phosphorescence Chemosensor for Homocysteine Based on an Iridium(III) Complex. Inorg. Chem. 46, 11075–11081 (2007).
Xiong, L. et al. Phosphorescence Imaging of Homocysteine and Cysteine in Living Cells Based on a Cationic Iridium(III) Complex. Inorg. Chem. 49, 6402–6408 (2010).
Wu, Y. et al. Ratiometric Phosphorescence Imaging of Hg(II) in Living Cells Based on a Neutral Iridium(III) Complex. Inorg. Chem. 50, 7412–7420 (2011).
Lu, L. et al. Detection of nicking endonuclease activity using a G-quadruplex-selective luminescent switch-on probe. Chem. Sci. 10.1039/C4SC02032D. (2014).
Wang, M. et al. A G-quadruplex-based, label-free, switch-on luminescent detection assay for Ag+ ions based on the exonuclease III-mediated digestion of C-Ag+-C DNA. J. Mater. Chem. B 2, 6467-6471 (2014).
Ma, D.-L. et al. Visualization of Zn2+ Ions in Live Zebrafish Using a Luminescent Iridium(III) Chemosensor. ACS Appl. Mater. Interfaces 6, 14008-14015 (2014).
Leung, K.-H. et al. Label-Free Luminescent Switch-on Detection of Endonuclease IV Activity Using a G-Quadruplex-Selective Iridium(III) Complex. ACS Appl. Mater. Interfaces 5, 12249-12253 (2013).
He, H.-Z., Chan, D. S.-H., Leung, C.-H. & Ma, D.-L. A highly selective G-quadruplex-based luminescent switch-on probe for the detection of gene deletion. Chem. Commun. 48, 9462-9464 (2012).
He, H.-Z. et al. Label-free luminescence switch-on detection of T4 polynucleotide kinase activity using a G-quadruplex-selective probe. Chem. Commun. 50, 5313-5315 (2014).
Leung, K.-H. et al. A luminescent G-quadruplex switch-on probe for the highly selective and tunable detection of cysteine and glutathione. Chem. Commun. 49, 771-773 (2013).
Kim, H.-B., Li, Y. & Hyun, M. H. Phosphorescent Chemosensor Based on Iridium(III) Complex for the Selective Detection of Cu(II) Ion in Aqueous Acetonitrile. Bull. Korean Chem. Soc. 23, 653 (2013).
Burns, D. T., Danzer, K. & Townshend, A. Use of the term “recovery” and “apparent recovery” in analytical procedures. Pure Appl. Chem. 74, 2201–2205 (2002).
Herrero, C. et al. Intramolecular light induced activation of a Salen-MnIII complex by a ruthenium photosensitizer. Chem. Commun. 46, 7605–7607 (2010).
Leung, K.-H. et al. Detection of base excision repair enzyme activity using a luminescent G-quadruplex selective switch-on probe. Chem. Commun. 49, 5630–5632 (2013).
Liang, Z., Liu, Z., Jiang, L. & Gao, Y. A new fluorescent chemosensor for copper(II) and molecular switch controlled by light. Tetrahedron Lett. 48, 1629–1632 (2007).
Kumar, M., Kumar, N., Bhalla, V., Sharma, P. R. & Kaur, T. Highly Selective Fluorescence Turn-on Chemodosimeter Based on Rhodamine for Nanomolar Detection of Copper Ions. Org. Lett. 14, 406–409 (2011).
Acknowledgements
This work is supported by Hong Kong Baptist University (FRG2/13-14/008), Centre for Cancer and Inflammation Research, School of Chinese Medicine (CCIR-SCM, HKBU), the Health and Medical Research Fund (HMRF/13121482), the Research Grants Council (HKBU/201811, HKBU/204612 and HKBU/201913), the French National Research Agency/Research Grants Council Joint Research Scheme (A-HKBU201/12), the State Key Laboratory of Environmental and Biological Analysis and Strategic Development Fund of HKBU (SKLP-14-15-P001), the Science and Technology Development Fund, Macao SAR (103/2012/A3) and the University of Macau (MYRG091(Y3-L2)-ICMS12-LCH, MYRG121(Y3-L2)-ICMS12-LCH and MRG023/LCH/2013/ICMS).
Author information
Authors and Affiliations
Contributions
M.W., K.H.L., S.L. and D.S.H.C. carried out all the experiments, performed the data analysis and wrote the manuscript. D.L.M. and C.H.L. designed the experiments and analyzed the results. D.W.J.K. analyzed the results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supporting Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Wang, M., Leung, KH., Lin, S. et al. A colorimetric chemosensor for Cu2+ ion detection based on an iridium(III) complex. Sci Rep 4, 6794 (2014). https://doi.org/10.1038/srep06794
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06794
This article is cited by
-
A Reversible Fluorescent Chemosensor for the Selective Detection of Cu2+ and CN- ions by Displacement Approach
Journal of Fluorescence (2023)
-
Determination of Cu2+ in drinking water using a hydroxyjulolidine-dihydroperimidine colorimetric sensor
Journal of Inclusion Phenomena and Macrocyclic Chemistry (2019)
-
C-Phycoerythrin as a Colorimetric and Fluorometric Probe for the Sensitive, Selective and Quantitative Detection of Cu2+ in Aqueous Samples
Journal of Fluorescence (2018)
-
Facile Green Synthesis of Gold Nanoparticles with Carboxymethyl Gum Karaya, Selective and Sensitive Colorimetric Detection of Copper (II) Ions
Journal of Cluster Science (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
