
Abstract
While biogeochemical and physical processes in the Southern Ocean are thought to be central to atmospheric CO2 rise during the last deglaciation, the role of the equatorial Pacific, where the largest CO2 source exists at present, remains largely unconstrained. Here we present seawater pH and pCO2 variations from fossil Porites corals in the mid equatorial Pacific offshore Tahiti based on a newly calibrated boron isotope paleo-pH proxy. Our new data, together with recalibrated existing data, indicate that a significant pCO2 increase (pH decrease), accompanied by anomalously large marine 14C reservoir ages, occurred following not only the Younger Dryas, but also Heinrich Stadial 1. These findings indicate an expanded zone of equatorial upwelling and resultant CO2 emission, which may be derived from higher subsurface dissolved inorganic carbon concentration.
Similar content being viewed by others

Introduction
Understanding the past condition of the surface ocean carbonate system and air-sea CO2 exchange is crucial to projecting future changes in the carbon cycle under ongoing anthropogenic global warming and ocean acidification. Atmospheric CO2 concentration increased by as much as 80 μatm during the last deglaciation, with ~50 μatm released during Heinrich Stadial 1 (HS1, from 17.5 to 14.6 kyr), followed by an additional ~30 μatm during the Younger Dryas (YD, from 12.9 to 11.7 kyr) (ref. 1). While the Southern Ocean is generally considered to be central to the deglacial CO2 rise2,3,4,5,6,7,8,9, the contribution from other oceanic regions remains relatively uninvestigated10,11,12,13. Information on the partial pressure of CO2 (pCO2) is needed to directly constrain past air-sea CO2 exchange and this can be reconstructed from boron isotopes (δ11B), a marine carbonate pH proxy14,15. Regions where surface seawater CO2 is out of equilibrium with the atmosphere are ideal for such studies and the equatorial Pacific is particularly well suited because it represents the largest global CO2 source at present (e.g. ref. 16). Corals broadly distributed in tropical to subtropical areas constitute excellent high-resolution geochemical archives for paleo-CO2 studies because they may be precisely radiogenically dated (U-series)3,17, unlike foraminifers that are affected by uncertainty in radiocarbon reservoir age (R).
Two previous studies have attempted to constrain equatorial Pacific CO2 changes. Palmer and Pearson10 showed increased CO2 emission during the last deglaciation in the western equatorial Pacific (WEP) from δ11B measurements on the planktonic foraminifer (Globigerinoides sacculifer) in a sediment core recovered from offshore Papua New Guinea (ERDC-92, Fig. 1a). Further east, Douville et al.11 performed δ11B on fossil corals from the Marquesas (9.5°S 139.4°W, Fig. 1a) and also demonstrated increased CO2 release at the end of the YD. However Douville et al.11 did not observe a significant CO2 release during HS1, complicating interpretation of the equatorial contribution to deglacial atmospheric CO2 rise. Integrated Ocean Drilling Program Expedition 310 (IODP Exp. 310)18 drilled the outer reef slope at Tahiti (17.6°S 149.5°W, Fig. 1a) recovering fossil corals from an open ocean environment spanning HS1, which enable us to assess the issue.

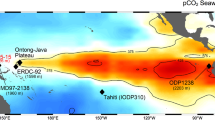
Sea surface pCO2 in the equatorial Pacific and vertical sections of the South Pacific.
(a) Equatorial Pacific locations discussed in the text with sea surface pCO2 for the reference year AD 2000 in which red and blue represent CO2 sources and sinks, respectively16. Vertical sections from 60°S to 30°N of (b) oxygen, (c) silicate, (d) DIC and (e) Δ14C. All data are downloaded and plotted using Ocean Data View software, version 4.5.5 (ref. 60). Inverted black triangles show locations of Tahiti and Marquesas. The white star and diamond in e indicate the locations of cores MV99-MC99/GC31/PC08 (refs. 36,43) and V21–31 (ref. 42), respectively.
Results
δ11B-pH calibration
This study establishes a new empirical δ11B-pH calibration utilizing, for the first time, anthropogenic ocean acidification. Empirical calibration is needed to overcome the observed offsets from a theoretical δ11B-pH curve in culture experiments for zooxanthellate corals14,15. (pH is reported using the total hydrogen scale, hereafter pH for simplicity). There are two primary approaches to overcoming the reported offsets. One is an empirical approach14 that assumes constant offsets in measured and theoretical δ11B (“offset a;” see Methods), while the other is an observational approach15 that considers potential pH modification by calcifiers.
The potential for pH-modification is of great concern for δ11B-based reconstruction of pH due to the implications for atmospheric pCO2 calculation. Such a phenomenon is consistent with indirect pH measurements of internal calcification fluid using pH sensitive dye that suggests a higher pH than ambient seawater, creating better conditions for calcification19. A ‘ΔpH’ concept in the δ11B-pH calibration that reflects pH differences in seawater (pHSW) and internal calcification fluid has been proposed15, however usage of this proposed relationship to calibrate δ11B of modern Porites spp. from Tahiti and Marquesas resulted in unrealistically high values (e.g. ~8.34 in AD 1991), well above reported estimates (e.g. refs. 20,21). Therefore the present study employs the empirical equation14 here (see Methods).
Ocean acidification was estimated from a combination of in situ fCO2 values in the surface ocean, atmospheric CO2 concentration directly measured at the Mauna Loa observatory in Hawaii since AD 1960 (ref. 22) and CO2 concentrations within bubbles trapped in an Antarctic ice core23 (Figs. 2, S1–S4; Supplementary Methods). These data were then fit to the previously reported δ11B measurements of Porites spp. (refs. 11,24), which are for the years AD 1991, 1950 and 1700 (Fig. 2, see Methods and Supplementary Methods for details of the δ11B-pH calibration and pH estimation since the Industrial Revolution).

Measured pCO2 from Mauna Loa22 and Law Dome ice core23 (red and blue lines) and estimated pH variation at Tahiti (black line).
Green triangles are newly calibrated pH from previously reported δ11B of modern corals from Tahiti and Marquesas11,24 (Table S1). Estimated pH from the AD 1700 Marquesas coral was scaled by 0.04 to correct for offset from Tahitian coral values. Error bars are 2σ.
pH and pCO2 reconstruction
Using our revised calibration, we reconstructed pH from our new δ11B measurements on Tahitian corals, as well as from previously reported data11 from both the Marquesas and Tahiti and the overall result is consistent with the WEP foraminifer δ11B variations10 (Fig 3a and b). The oldest coral sample, dated to 20.7 ka BP during the last glacial maximum (LGM), exhibits a relatively high pH (8.26). From 15.5 to 9.0 ka BP, pH is generally constant within uncertainty (8.15–8.22) and consistent with the preindustrial value of 8.20. Four notable pH excursions are associated with HS1 and the YD. Two of our samples exhibit anomalously low pH at the end of HS1 (8.13 at 15.15 ka and 8.09 at 14.99 ka BP), in addition to those at end of the YD at the Marquesas11. The low pH following HS1 had been previously undetected at this location. Calculation of pCO2 (see Methods) reveals deglacial values significantly above those of the atmosphere (Figs. 3c and 4a). Conversely, ΔpCO2 during last glacial and the early Holocene was nearly zero, suggesting air-sea CO2 equilibrium.

Deglacial δ11B, pH and pCO2 variability in the equatorial Pacific.
(a) Reported δ11B values of planktonic foraminifera with 2σ uncertainty from ERDC-92 (ref. 10). Age model is from the original publication. (b) In situ pH reconstructed from δ11B of fossil Porites spp. using our new calibration. Red and green circles are from Tahiti by this study and Douville et al.11, respectively. Blue circles are from Marquesas by Douville et al.11 after correction by +0.04 pH units. (c) Derived pCO2 of surface water around equatorial South Pacific Ocean (same colors as b) and atmospheric pCO2 on the GICC05 timescale1. All error bars are 2σ. YD, Younger Dryas; B/A, Bølling/Allerød; ACR, Antarctic Cold Reversal; HS1, Heinrich Stadial 1; LGM, Last Glacial Maximum.

Differences between modern and deglacial pCO2 and R (ΔpCO2 and Rdiff).
The last deglaciation is characterized by larger and more variable pCO2 and R values. (a) Atmosphere-ocean ΔpCO2 based on Tahiti and Marquesas corals and Antarctic ice core1. Figure legend is same as Fig. 3. (b) Rdiff around the equatorial Pacific Ocean calculated from fossil corals (red: offshore Tahiti; orange: reef crest of Tahiti barrier reef58, blue: Marquesas26; light blue: Kiritimati59; black: Mururoa58). Horizontal dashed lines represent ΔpCO2 = 0 and Rdiff = 0.
Results from a different portion of the same 14.99 ka BP coral sample deviate by as much as 1.4‰, which corresponds to 0.11 in pH and 100 μatm in pCO2 (310-M0024A-11R-1W_77-90 and 310-M0024A-11R-1W_60-75, Table S1). Considering the average ~4 year temporal resolution of each sample, these excursions occurred abruptly and persisted for several years, which differs from modern observations that show no clear interannual or decadal variability (Supplementary Methods, Fig. S1). This enhanced variability, which is also observed in Sr/Ca derived SST results from another Porites colony recovered from IODP Exp. 310 (ref. 25), may relate to Tahiti's location at the rim of equatorial upwelling cell (Fig. 1a). Taken together, pCO2 (pH) records indicate that the equatorial Pacific became a larger CO2 source during the last deglaciation with excursions at the end of HS1 and the YD.
Marine 14C reservoir age compilation
Compiled marine 14C reservoir ages (R) throughout the equatorial Pacific resemble pCO2 variability (see Methods; Figs. 4 and S5; Table S2). Larger and more variable values of R are evident in Tahiti during HS1 and the YD and enhanced R variability is also seen in the Marquesas (Figs. 4 and S5). Paterne et al.26 sub-sampled different parts in the same fossil coral skeleton and analyzed both 14C and U/Th. They observed no difference in U/Th dates, but a much larger difference in 14C. Possibilities of either a diagenetic alteration or a change in R were suggested. The latter is more probable because a large variation in R is also suggested from Vanuatu coral at 11.7–12.4 ka (~400 years; during the YD, ref. 27).
Though reported Rdiff (difference between calculated R and modern R; see Methods) data around the upwelling zone during the LGM are sparse, calculations with the new Lake Suigetsu datasets28 suggests no substantial change in R (see Methods; Figs. 4 and S5). This implies that the CO2 exchange rate in the surface equatorial Pacific during the last glacial was almost the same as present, which supports the above-mentioned observation that ΔpCO2 is essentially equivalent to zero and indicates that anomalous R values are limited to the last deglaciation.
Discussion
pCO2 variability in subtropical oligotrophic water can be explained by mixing of water masses that exhibit distinctly different dissolved inorganic carbon (DIC) concentrations. A southward migration of the intertropical convergence zone (ITCZ) that partly controls thermocline depth is hypothesized during Heinrich Events including HS1 and the YD (e.g. ref. 29). At present, the ITCZ does not seem to affect surface pCO2 variability (Fig. 1a) and if it is displaced southward, the locus of equatorial upwelling remains at the equator due to the influence of inter-hemispheric asymmetry of Coriolis force (e.g. refs. 30,31). Enhanced upwelling (shallower thermocline, La Niña-like conditions) or increased subsurface DIC concentration are more likely to drive pCO2 variability based on sedimentary evidence from the equatorial Pacific for higher nutrient content, e.g., enhanced biogenic opal export production and lower stable carbon isotopes (δ13C) (TT013-PC72, ODP Site 1240 and TR163-19)32,33,34,35 (Fig. 1a). Semi-conservative radiogenic neodymium isotopes (εNd) from sediment cores at the eastern equatorial Pacific (EEP) (ODP Site 1240) and off Baja California (MV99-MC99-GC31/PC08) indicate stronger subsurface water transport from the south33,36 (Fig. 1). Covariation of geochemical properties between the Southern Ocean and the equatorial Pacific suggest a subsurface connection during the last deglaciation (e.g. refs. 32,33,34,35,36,37,38). Thus, pCO2 variability may be explained by an increase in DIC in the upwelled, subsurface water masses as opposed to physical processes alone.
Water mass subduction along the subantarctic front, mainly off Chile39, forms Subantarctic Mode Water (SAMW) and Antarctic Intermediate Water (AAIW) that upwells at the equatorial Pacific via the Equatorial Undercurrent (EUC) (Fig. 1). SAMW and AAIW are characterized by higher/lower concentrations of oxygen/silicic acid (Fig. 1b,c). It is suggested that the abyssal DIC reservoir around the Southern Ocean increased during the last glacial period6,7,8,40, which would have contributed to lower atmospheric pCO2. Carbon dioxide was released to the surface through deep ocean ventilation during HS1 and the YD (refs. 2,3,4,5,6,7,8,9), however export production was insufficient to fully compensate the increased carbon flux41. This is consistent with residual radiocarbon content (Δ14C) of intermediate water at the EEP (V21–30)42 and off Baja California (MV99-MC99-GC31/PC08)43 that indicates anomalously older water was incorporated into SAMW/AAIW (Fig. 1), as well as with depleted δ13C of surface and lower thermocline dwelling foraminifers from sediment cores at both equatorial (TT013-PC72, ODP Site 1240 and TR163-19) and South Pacific sites32,33,34,35,37,38. Moreover, enhanced export production of biogenic opal suggest more silicic acid was transported via the EUC to thermocline water at the equatorial Pacific (V19-30 and TT013-PC72) without being consumed completely within the Southern Ocean4,35,41. Stronger Ekman transport in association with sea ice retreat and a poleward shift of southern westeries is suggested to be a driver4,5.
A similarity between R and pCO2 variability during the last deglaciation supports an interpretation that older DIC was incorporated to subtropical surface water through mixing with SAMW/AAIW, though, contrary evidence comes from the current formation sites off Chile44 and New Zealand6. However, a key sediment record off Chile was recently reevaluated and the new interpretations indicate stronger upwelling and subsequent larger R in surface water in the Southern Ocean9, which agrees well with our interpretation. Yet, further work is still needed to fully understand both the physical and biogeochemical dynamics in the Southern Ocean and the equatorial Pacific2.
Positive ΔpCO2 indicates CO2 flux from the ocean to the atmosphere. Previous studies10,11 indicated that the equatorial Pacific contributed to deglacial CO2 rise, however the timing of anomalously higher pCO2 events recorded in radiogenically dated fossil corals do not systematically correspond to those of atmospheric CO2 rise recovered from Antarctic ice core on the GICC05 timescale1 (Figs. 3 and 4). Moreover our new calibration reveals a modest CO2 emission continued through the Bølling/Allerød/Antarctic Cold Reversal when no atmospheric CO2 increase is observed (Figs. 3 and 4). Though we demonstrate that the equatorial Pacific became a larger CO2 source during the last deglaciation, it is too early to conclude its exact contribution to atmospheric CO2 rise. The Southern Ocean is suggested to be central in CO2 degassing4,5,6,7,8,9 and the contribution of the terrestrial biosphere should be further evaluated45. More evidence spanning the YD and the early part of HS1, in particular the sharp rise in atmospheric CO2 and the sudden drop of δ13C of CO2 (refs. 1,46), as well as more spatial coverage is needed.
Methods
δ11B analyses
The δ11B values of fossil Porites spp. were measured following the protocol of Ishikawa and Nagaishi47. Fossil corals were screened for diagenetic alteration with X-ray diffraction and geochemical analyses, as well as visual using a Scanning Electron Microscope48. Bulk sampling was conducted along the growth axis and time resolution of each sample is several years (1–8 years) depending on growth rate of each coral48. Typically 6 mg of carbonate was used for δ11B measurement. After removals of organic matter using 30% H2O2 for ~12 hours, boron was purified by cation and anion exchange using AG 50 W X12 and 1-X4 resin (Bio-Rad, USA) and then δ11B were measured using the positive polarity thermal ionization mass spectrometer (P-TIMS; Thermo Finnigan TRITON) installed at Kochi Core Center, Japan Agency for Marine-Earth Science and Technology. All reported δ11B values are the mean of duplicate analyses (Table S1). Repeated analysis of the JCp-1, carbonate standard provided by Geological Survey of Japan yielded 24.21 ± 0.18‰ (2σ, n = 18), which is the finest precision to date47. Differences between the duplicates are 0.08‰ on average with the largest one of 0.28‰ (Table S1), which is within the measurement uncertainty of JCp-1. We conservatively report ±0.18‰ as the analytical uncertainty of our δ11B measurements.
δ11B-pH calibration and pCO2 calculation
First, the analytical procedure-specific isotopic offset49 was corrected using the equation bellow (modified after Zeebe & Wolf-Gladrow50) in order to reduce inter-laboratory offsets in reported δ11B values,

where δ11Bcarbonate-corrected is the boron isotopic value of carbonate after correction and δ11BSW-stacked is the global average δ11B of seawater. We used the recommended value of 39.61‰ for δ11BSW-stacked (ref. 49). δ11Bcarbonate-measured and δ11BSW-measured are δ11B of carbonate samples and of seawater measured in different laboratories. Without performing inter-laboratory correction of JCp-1, results of Douville et al.11 and this study differ by 0.25‰, equivalent to ~0.03 pH units. After the correction using the reported value of δ11BSW-measured (refs. 11,47), the difference improved to be 0.07‰, indicating that correction should be performed.
After the correction, the empirical calibration equation reported by Hönisch et al.14 was utilized. In this equation, the vital offset ‘a’ and fractionation factor ‘α3-4’ that yield the lowest erms (root-mean square error) is determined.

Three previously reported δ11B values for modern Porites spp. from Tahiti and Marquesas were fit to estimated pH at AD 1991, 1950 and 1700 (refs. 11,24) (Fig. 2, Table S1). In the calibration ‘a’ was determined as ‘−6.0’ when we chose recently reevaluated α3-4 of ‘1.0272’ (ref. 51). When this calibration was conducted, both ‘a’ and ‘α3-4’ were allowed to vary freely and the resultant α3-4 with the lowest erms was very close to that of Klochko et al.51 rather than the previously accepted value of Kakihana et al.52 Thus we used Klochko's fractionation factor. pH was calculated from both previously reported and newly obtained δ11B values considering a local difference of seawater pH between Tahiti and Marquesas (see Supplementary Methods for details).
pCO2 was further calculated from obtained pH values from δ11Bcarbonate-corrected using CO2SYS program under assumptions of constant temperature, salinity and total alkalinity (see also Supplementary Methods for details). The δ11B values of G. sacclifer10 are not included here due to large uncertainties in the δ11B-pH calibration (e.g. refs. 12,15).
R compilation
Published 14C (radiocarbon years) and U/Th ages of fossil coral samples obtained during IODP Exp. 310 were compiled in order to calculate residual radiocarbon activities (Δ14C) and R. We verified via IODP sample ID and core photographs18 that the exact same samples were selected (Table S2). In some cases different portion of the skeleton of the same coral was dated. Given that lifetimes of coral are generally less than several decades, temporal gaps derived from sub-sampling are negligible in calculations of Δ14C and R. We did not use 14C ages from either microbialite (carbonate created by bacteria) or encrusting coralline algae from equivalent down-core depths due to a possibility of post-depositional growth (for details, see ref. 53). Calculation was done according to equations (3) and (4) where 14C-age is an original radiocarbon data before a local R correction54,55.


Atmospheric Δ14C (Δ14Catm) from INTCAL09 (ref. 56) was used to calculate R. Samples that span 29–30 ka BP were calculated using the recently published Lake Suigetsu Δ14Catm dataset28, since as much as ~100‰ offsets are observed between INTCAL09 in this interval (Fig. S5a). Southon et al.57 points out that the INTCAL09 curve heavily relies on Cariaco Basin varve sediment data beyond the dendro-chronological limit (>12.55 ka BP) and has deficits during the YD and HS1 due to changes in local R in the equatorial Atlantic. Specifically, a difference during 15.5–17.0 ka BP is apparent, however it does not affect our calculation since there are no samples that span this interval (Fig. S5). Other 14C and U/Th datasets for fossil corals from the equatorial Pacific islands were also used to calculate R including Tahiti58, Marquesas26, Kiritimati59 and Mururoa58 (Figs. 1, 4 and S5b). Rdiff denotes differences between calculated R and modern R (235 ± 110 for Tahiti56, 390 ± 60 for Marquesas26, 335 ± 100 for Kiritimati56, 300 ± 100 for Mururoa58), thus it differs from ΔR that conventionally represents local 14C reservoir age. We estimate that the accumulated uncertainty in the Rdiff calculation are the sum of errors in 14C dating, U/Th dating and modern R (Fig. S5b; Table S2).
References
Lourantou, A. et al. Constraint of the CO2 rise by new atmospheric carbon isotopic measurements during the last deglaciation. Glob. Biogeochem. Cycles 24, GB003545 (2010).
Matsumoto, K. & Yokoyama, Y. Atmospheric Δ14C reduction in simulations of Atlantic overturning circulation shutdown. Glob. Biogeochem. Cycles 27, 296–304 (2013).
Yokoyama, Y. & Esat, T. M. Global climate and sea level- enduring variability and rapid fluctuations over the past 150,000 years. Oceanography 24, 54–69 (2011).
Anderson, R. F. et al. Wind-Driven Upwelling in the Southern Ocean and the Deglacial Rise in Atmospheric CO2 . Science 323, 1443–1448 (2009).
Toggweiler, J. R., Russel, J. L. & Carson, S. R. Midlatitude westerlies, atmospheric CO2 and climate change during the ice ages. Paleoceanography 21, PA2005 (2006).
Rose, K. A. et al. Upper-ocean-to-atmosphere radiocarbon offsets imply fast deglacial carbon dioxide release. Nature 466, 1093–1097 (2010).
Burke, A. & Robinson, L. F. The Southern Ocean's Role in Carbon Exchange During the Last Deglaciation. Science 335, 557–561 (2012).
Skinner, L. C., Fallon, S., Waelbroeck, C., Michel, E. & Barker, S. Ventilation of the Deep Southern Ocean and Deglacial CO2 Rise. Science 328, 1147–1151 (2010).
Siani, G. et al. Carbon isotope records reveal precise timing of enhanced Southern Ocean upwelling during the last deglaciation. Nat. Commun. 4:2758, 10.1038/ncomms3758 (2013).
Palmer, M. R. & Pearson, P. N. A 23,000-Year Record of Surface Water pH and pCO2 in the Western Equatorial Pacific Ocean. Science 300, 480–482 (2003).
Douville, E. et al. Abrupt sea surface pH change at the end of the Younger Dryas in the central sub-equatorial Pacific inferred from boron isotope abundance in corals (Porites). Biogeosciences 7, 2445–2459 (2010).
Henehan, M. J. et al. Calibration of the boron isotope proxy in the planktonic foraminifera Globigerinoides ruber for use in palaeo-CO2 reconstruction. Earth Planet. Sci. Lett. 364, 111–122 (2013).
Yu, J., Thornalley, D. J. R., Rae, J. W. B. & McCave, N. I. Calibration and application of B/Ca, Cd/Ca and δ11B in Neogloboquadrina pachyderma (sinistral) to constrain CO2 uptake in the subpolar North Atlantic during the last deglaciation. Paleoceanography 28, 237–252 (2013).
Hönisch, B., Hemming, N. G. & Loose, B. Comment on ‘A critical evaluation of the boron isotope-pH proxy: the accuracy of ancient ocean pH estimates’ by M. Pagani, D. Lemarchand, A. Spivak, J. Gaillardet. Geochim. Cosmochim. Acta 71, 1636–1641 (2007).
Trotter, J. et al. Quantifying the pH ‘vital effect’ in the temperate zooxanthellate coral Cladocora caespitosa: Validation of the boron seawater pH proxy. Earth Planet. Sci. Lett. 303, 163–173 (2011).
Takahashi, T. et al. Climatological mean and decadal change in surface ocean pCO2 and net sea–air CO2 flux over the global oceans. Deep-Sea Res. II 56, 554–577 (2009).
Deschamps, P. et al. Ice-sheet collapse and sea-level rise at the Bølling warming 14,600 years ago. Nature 483, 559–564 (2012).
Camoin, G. F., Iryu, Y., McInroy, D. & the IODP Expedition 310 Scientists. IODP Expedition 310 Reconstructs Sea Level, Climatic and Environmental Changes in the South Pacific during the Last Deglaciation. Sci. Drill. 5, 4–12 (2007).
Venn, A. A. et al. Impact of seawater acidification on pH at the tissue–skeleton interface and calcification in reef corals. Proc. Natl. Acad. Sci. USA 110, 1634–1639 (2013).
Caldeira, K. & Wickett, M. E. Anthropogenic carbon and ocean pH. Nature 425, 365 (2003).
Key, R. M. et al. A global ocean carbon climatology: Results from Global Data Analysis Project (GLODAP). Glob. Biogeochem. Cycle 18, GB002247 (2004).
Keeling, R. F., Piper, S. C., Bollenbacher, A. F. & Walker, J. S. Atmospheric CO2 records from sites in the SIO air sampling network. In Trends: A Compendium of Data on Global Change. Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge, Tenn., U.S.A. 10.3334/CDIAC/atg.035 (2009).
Etheridge, D. M. et al. Natural and anthropogenic changes in atmospheric CO2 over the last 1000 years from air in Antarctic ice and firn. J. Geophys. Res. 101, 4115–4128 (1996).
Gaillardet, J. & Allègre, C. J. Boron isotopic compositions of corals: seawater or diagenesis record? Earth Planet. Sci. Lett. 136, 665–676 (1995).
Felis, T. et al. Pronounced interannual variability in tropical South Pacific temperatures during Heinrich Stadial 1. Nat. Commun. 3:965, 10.1038/ncomms1973 (2012).
Paterne, M. et al. Paired 14C and 230Th/U dating of surface corals from the Marquesas and Vanuatu (sub-equatorial Pacific) in the 3000 to 15,000 cal yr interval. Radiocarbon 46, 551–566 (2004).
Burr, G. S. et al. A high-resolution radiocarbon calibration between 11,700 and 12,400 calendar years BP derived from 230Th ages of corals from Espiritu Santo island, Vanuatu. Radiocarbon 40, 1093–1105 (1998).
Bronk-Ramsey, C. et al. A Complete Terrestrial Radiocarbon Record for 11.2 to 52.8 kyr B.P. Science 338, 370–374 (2012).
Deplazes, G. et al. Links between tropical rainfall and North Atlantic climate during the last glacial period. Nature Geosci. 6, 213–217 (2013).
Talley, L. D., Pickard, G. L., Emery, W. J. & Swift, J. H. (2011) in Descriptive Physical Oceanography: An Introduction.(Elsevier Academic Press, Amsterdam).
Sarmiento, J. L. & Gruber, N. in Ocean Biogeochemical Dynamics. (Prinston Univ. Press, 2011).
Spero, H. J. & Lea, D. W. The Cause of Carbon Isotope Minimum Events on Glacial Terminations. Science 296, 522–525 (2002).
Pena, L. D. et al. Rapid changes in meridional advection of Southern Ocean intermediate waters to the tropical Pacific during the last 30 kyr. Earth Planet. Sci. Lett. 368, 20–32 (2013).
Pena, L. D., Cacho, I., Ferretti, P. & Hall, M. A. El Nino–Southern Oscillation–like variability during glacial terminations and interlatitudinal teleconnections. Paleoceanography 23, PA001620 (2008).
Hayes, C. T., Anderson, R. F. & Fleisher, M. Q. Opal accumulation rates in the equatorial Pacific and mechanisms of deglaciation. Paleoceanography 26, PA002008 (2011).
Basak, C., Martin, E. E., Horikawa, K. & Marchitto, T. M. Southern Ocean source of 14C-depleted carbon in the North Pacific Ocean during the last deglaciation. Nature Geosci. 3, 770–773 (2010).
Loubere, P. & Bennet, S. Southern Ocean biogeochemical impact on the tropical ocean: Stable isotope records from the Pacific for the past 25,000 years. Glob. Planet. Change 63, 333–340 (2008).
Bostock, H. C., Opdyke, B. N., Gagan, M. K. & Fifield, L. K. Carbon isotope evidence for changes in Antarctic Intermediate Water circulation and ocean ventilation in the southwest Pacific during the last deglaciation. Paleoceanography 19, PA001047 (2004).
Bostock, H. C., Sutton, P. J., Williams, M. J. M. & Opdyke, B. N. Reviewing the circulation and mixing of Antarctic Intermediate Water in the South Pacific using evidence from geochemical tracers and Argo float trajectories. Deep-Sea Res. I 73, 84–98 (2013).
Sikes, E. L., Samson, C. R., Guilderson, T. P. & Howard, W. R. Old radiocarbon ages in the southwest Pacific Ocean during the last glacial period and deglaciation. Nature 405, 555–559 (2000).
Horn, M. G., Beucher, C. P., Robinson, R. S. & Brezeyinski, M. A. Southern ocean nitrogen and silicon dynamics during the last deglaciation. Earth Planet. Sci. Lett. 310, 334–339 (2011).
Stott, L., Southon, J., Timmermann, A. & Koutavas, A. Radiocarbon age anomaly at intermediate water depth in the Pacific Ocean during the last deglaciation. Paleoceanography 24, PA001690 (2009).
Marchitto, T. M., Lehman, S. J., Ortiz, J. D., Flückiger, J. & van Green, A. Marine Radiocarbon Evidence for the Mechanism of Deglacial Atmospheric CO2 Rise. Science 316, 1456–1459 (2007).
De Pol-Holz, R., Keigwin, L., Southon, J., Hebbelin, D. & Mohtadi, M. No signature of abyssal carbon in intermediate waters off Chile during deglaciation. Nature Geosci. 3, 192–195 (2010).
Ciais, P. et al. (2012) Large inert carbon pool in the terrestrial biosphere during the Last Glacial Maximum. Nature Geosci 5, 74–79.
Schmitt, J. et al. Carbon Isotope Constraints on the Deglacial CO2 Rise from Ice Cores. Science 336, 711–714 (2012).
Ishikawa, T. & Nagaishi, K. High-precision isotopic analysis of boron by positive thermal ionization mass spectrometry with sample preheating. J. Anal. At. Spectrom. 26, 359–365 (2011).
Inoue, M. et al. Trace element variations in fossil corals from Tahiti collected by IODP Expedition 310: Reconstruction of marine environments during the last deglaciation (15 to 9 ka). Mar. Geol. 271, 303–306 (2010).
Foster, G. L., Pogge von Strandmann, P. A. E. & Rae, J. W. B. Boron and magnesium isotopic composition of seawater. Geochem. Geophys. Geosyst. 11, GC003201 (2010).
Zeebe, R. E. & Wolf-Gladrow, D. in CO2 in Seawater: Equilibrium, kinetics, isotopes. (Elsevier Oceanography Series, 2001).
Klochko, K. et al. Experimental measurement of boron isotope fractionation in seawater. Earth Planet. Sci. Lett. 248, 276–285 (2006).
Kakihana, H., Kotaka, M., Satoh, S., Nomura, M. & Okamoto, M. Fundamental Studies on the Ion-Exchange Separation of Boron Isotopes. Bull. Chem. Soc. Japan 50, 158–163 (1977).
Seard, C. et al. Microbialite development patterns in the last deglacial reefs from Tahiti (French Polynesia; IODP Expedition #310): Implications on reef framework architecture. Mar. Geol. 279, 63–86 (2011).
Adkins, J. F. & Boyle, E. A. Changing atmospheric Δ14C and the record of deepwater paleoventilation ages. Paleoceanography 12, 337–344 (1997).
Burr, G. S. et al. Modern and Pleistocene reservoir ages inferred from South Pacific Corals. Radiocarbon 51, 319–335 (2009).
Reimer, P. J. et al. IntCal09 and Marine09 radiocarbon age calibration curves, 0–50,000 years cal BP. Radiocarbon 51, 1111–1150 (2009).
Southon, J., Noronha, A. L., Cheng, H., Edwards, R. L. & Wang, Y. A high-resolution record of atmospheric 14C based on Hulu Cave speleothem H82. Quat. Sci. Rev. 33, 32–41 (2012).
Bard, E., Arnold, M., Hamelin, B., Tisnerat, L. N. & Cabioch, G. Radiocarbon calibration by means of mass spectrometric 230Th/234U and 14C ages of corals: an updated database including samples from Barbados, Mururoa and Tahiti. Radiocarbon 40, 1085–1092 (1998).
Fairbanks, R. G. et al. Radiocarbon calibration curve spanning 0 to 50,000 years BP based on paired 230Th/234U/238U and 14C dates on pristine corals. Quat. Sci. Rev. 24, 1781–1796 (2005).
Schlitzer, R. Ocean Data View. <http://odv.awi.de>, (2014) Date of access:29/01/2014.
Acknowledgements
We thank the IODP Exp. 310 Science Party, in particular G. Camoin, Y. Iryu and D. McInroy, as well as W. Hale, U. Röhl, A. Wülbers and all the staff of the Bremen Core Repository. We would like to thank J. Matsuoka and K. Nagaishi for technical support. Our gratitude is also extended to M. Inoue, Y. Miyairi, M. Becchaku and S. Yoshinaga for laboratory assistance. This study was partly supported by the JSPS NEXT program GR031 and grant number 26247085 to Y.Y., grant number 24340127 to T.I. and Research fellowships for young scientists to K.K.
Author information
Authors and Affiliations
Contributions
Y.Y. designed the study and obtained samples. K.K. and T.I. measured coral boron isotope ratios. A.S. prepared samples. S.O. performed the statistical analysis of the data. K.K., Y.Y., T.I., S.O. and A.S. contributed to the interpretation and the preparation of the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kubota, K., Yokoyama, Y., Ishikawa, T. et al. Larger CO2 source at the equatorial Pacific during the last deglaciation. Sci Rep 4, 5261 (2014). https://doi.org/10.1038/srep05261
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05261
This article is cited by
-
Southern Ocean glacial conditions and their influence on deglacial events
Nature Reviews Earth & Environment (2023)
-
Deglacial restructuring of the Eastern equatorial Pacific oxygen minimum zone
Communications Earth & Environment (2022)
-
Recent ocean acidification trends from boron isotope (δ11B) records of coral: Role of oceanographic processes and anthropogenic CO2 forcing
Journal of Earth System Science (2022)
-
Equatorial Pacific seawater pCO2 variability since the last glacial period
Scientific Reports (2019)
-
An atmospheric chronology for the glacial-deglacial Eastern Equatorial Pacific
Nature Communications (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
