
Abstract
The ocean may have played a central role in the atmospheric pCO2 rise during the last deglaciation. However, evidence on where carbon was exchanged between the ocean and the atmosphere in this period is still lacking, hampering our understanding of global carbon cycle on glacial–interglacial timescales. Here we report a new surface seawater pCO2 reconstruction for the western equatorial Pacific Ocean based on boron isotope analysis—a seawater pCO2 proxy—using two species of near-surface dwelling foraminifera from the same marine sediment core. The results indicate that the region remained a modest CO2 sink throughout the last deglaciation.
Similar content being viewed by others
Introduction
During the last deglaciation (ca. 19.0–10.5 ka), the atmospheric partial pressure of carbon dioxide (pCO2) increased by approximately 80 μatm1,2,3. Lines of evidence suggest that the deep-ocean carbon reservoir played a central role in the rise in atmospheric CO2, since its carbon storage capacity is 60 times larger than that of the atmosphere4,5,6,7,8. However, evidence on where carbon was exchanged between the ocean and the atmosphere is still lacking, hampering our understanding of global carbon cycle on glacial–interglacial timescales.
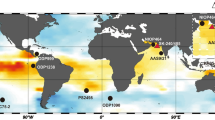
The boron isotope ratio (δ11B) of the skeletons of marine calcifying organisms such as corals and foraminifera can be used to infer where a CO2 source or sink existed, because the δ11B value of marine calcium carbonates is dependent on seawater pH, from which the pCO2 of the seawater can be reconstructed9,10,11,12. Investigating current oceanic source regions of CO2, such as the equatorial Pacific, is of great importance because such regions were likely sites of CO2 transfer from the deep-sea to the atmosphere in the past, as they are today (Fig. 1). In previous studies that measured δ11B in biogenic calcium carbonates, Martinez-Boti et al.10 (ODP Site 1238, Fig. 1) and Kubota et al.11 (IODP Exp. 310) revealed that central and eastern parts of the equatorial Pacific (CEP and EEP, respectively) acted as CO2 sources during the last deglaciation, suggesting that the equatorial Pacific ocean partly contributed to the rise in atmospheric CO2. However, some controversial results have been obtained in a marine sediment record from the western equatorial Pacific (WEP)12. Based on δ11B measurements of the planktonic foraminifera Torilobatus sacculifer in a marine sediment core (ERDC-92, Fig. 1), Palmer and Pearson12 reported that the surface layer of the WEP was a CO2 source during the last deglaciation, which suggested a potential contribution of this region to the deglacial rise in atmospheric CO2. However, their reported ranges of δ11B values differ from generally accepted values for T. sacculifer on glacial-interglacial timescales9,10,13,14,15,16 by as much as 5‰, which is likely due to positive bias, especially when analyzing foraminifera shell using negative thermal ionization mass spectrometry (N-TIMS)15,17,18,19. Considering that glacial–interglacial changes in δ11B measured in foraminifera are as small as approximately 2‰, this large discrepancy cannot be overlooked. The δ11B measurement of biogenic calcium carbonate using N-TIMS method has started since the end of 1980s (ref.20), and there has been a significant development in analytical procedure15,16,17,18,19. However, it still suffers from mass fractionation during ionization of the sample, resulting in a larger analytical uncertainty as well as less accuracy, especially when foraminifera sample is analyzed17,18,19. The δ11B measurement using multi-collector inductively coupled plasma mass spectrometry (MC-ICPMS) has started since the beginning of 2000s (refs21,22), which has become more widely-accepted and conventional ways to determine δ11B values, providing more precise and accurate values (e.g., refs9,10,11,13,14,15,16,17,18,19,23,24). Therefore, the refinement of seawater pCO2 records derived from the δ11B of planktonic foraminifera from the WEP, determined by MC-ICPMS method, is of great importance for understanding the role of the equatorial Pacific in the deglacial rise in atmospheric CO2. The WEP drives the world’s most intense atmospheric convection, being regarded as a heat engine of earth climate system25, thus to understand past oceanographic variability is of great importance not only for carbon cycle but also for climatology. For this purpose, we chose a marine sediment core recovered from the West Caroline Basin (KR05-15 PC01, 0.10°S 139.58°E, 3226 m below sea level, Fig. 1)26. A chronology of core KR05-15 PC01 was constructed based on 17 radiocarbon dates and deterministic age-depth modeling27, as well as oxygen isotope measurements of benthic foraminifera shells (Supplementary Fig. 1).

Sea surface pCO2 in the equatorial Pacific. Equatorial Pacific locations discussed in the text with sea surface pCO2 values for the reference year 2000 CE. Red and blue areas represent CO2 sources and sinks, respectively42. Diamonds indicate the locations of marine sediment cores and fossil corals mentioned in this study.
Boron Isotopes and Seawater CO2 System Reconstruction
In this study, we analyzed two species of surface-dwelling planktonic foraminifera—Globigerinoides ruber (white) and T. sacculifer. We collected foraminifera shells from size fractions generally used in paleoceanographic studies, namely, 300–355 μm and 500–855 μm, for G. ruber and T. sacculifer (with a sac-like final chamber), respectively9,10,13,14,15,16,24. To calculate pCO2, it is necessary to measure not only δ11B but also the Mg/Ca ratio (a temperature proxy) and oxygen isotope ratio (δ18O, a salinity proxy). Thus, we established a way to obtain these three values from a single sample, which requires 3–7 mg of foraminifera shells, from which shells are removed that potentially contain fragments of shells from other calcifying organisms and siliciclastic grains (Supplementary Figs S2–S7).
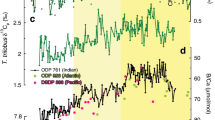
The δ11B values obtained in this study lie within the range obtained by Henehan et al.13 for both G. ruber and T. sacculifer, and are also consistent with the reported δ11B values from the core-top material of marine sediment collected from the Ontong Java Plateau in the WEP14 (Fig. 1, Supplementary Fig. S7). Both foraminifera records show steady δ11B values during the last glacial period (31–20 ka) (Fig. 2a), and both then show gradual decreases in δ11B, starting at the beginning of the last deglaciation, with the onset of the decrease in T. sacculifer preceding that in G. ruber by approximately 3 ky. Before 8 ka, the δ11B values of T. sacculifer are always lower than those of G. ruber, with a difference of 1.0–1.5‰. We estimate the calcification depth of G. ruber and T. sacculifer from theoretically predicted δ18O and δ11B values using data spanning the Holocene (4.0–10.5 ka). Since water temperature and pH (and pCO2) vary with water depth (Fig. 3a–c), the measured δ18O and δ11B values of foraminifera shells can be used to estimate the mean water depth at which the calcite shells precipitated. The theoretical δ18O depth profile of foraminifera calcite is calculated from temperature and seawater δ18O values. For the calculation of theoretical δ11B depth profile of both species, seawater pH is used, in which a decrease of pH due to anthropogenic CO2 incorporation since the Industrial Revolution is considered. The estimates of calcification depth based on different isotopes showed good agreement (Fig. 3d,e). Estimated calcification depths were 0–75 m (within the mixed layer) for G. ruber, and 75–125 m (below the mixed layer; upper thermocline) for T. sacculifer (Fig. 3), which is consistent with independent estimates from marine sediment cores collected in the WEP28,29,30. In the present study, T. sacculifer shells recorded colder, lower pH, and higher pCO2 water compared with G. ruber shells (Figs 2 and 3).

Deglacial changes of seawater CO2 system. Deglacial δ11B, pH, pCO2, and ΔpCO2 (seawater pCO2 minus atmospheric pCO2) variability in the equatorial Pacific. (a) δ11B values of G. ruber (red) and T. sacculifer (black) from core KR05-15 PC01 with 2σ analytical uncertainty. (b) pH (in total hydrogen scale) of surface and upper-thermocline seawater reconstructed from δ11B. (c) Reconstructed pCO2 of surface and upper-thermocline seawater and atmospheric pCO2 reconstructed from Antarctic ice cores (West Antarctic Ice Sheet Divide ice core, WDC; Siple Dome; EPICA Dome C, EDC)1,2,3. (d) Reconstructed pCO2 of surface seawater in the WEP (red, this study), EEP (light green)10, and CEP (dark green)11,23. Error bars of pH, pCO2, and ΔpCO2 were calculated using a Monte Carlo approach. The shaded area indicates the time period of the last deglaciation.

Depth profile of seawater CO2 system and calcification depth estimation of planktonic foraminifera. Depth profile of seawater properties and calcification depth estimation of G. ruber, and T. sacculifer (with sac-like final chamber), in the WEP. (a) Depth profile of water temperature74. The shaded area indicates thermocline depth. (b) Seawater pH and (c) seawater pCO2 calculated using pre-industrial dissolved inorganic carbon and total alkalinity43. (d) δ18O and (e) δ11B values estimated using water temperature, salinity, seawater pH, and species-specific δ11B–pH calibration equations.
Shell Dissolution Effect
Because our core was obtained from water below the calcite saturation horizon depth (the saturation state with respect to calcite, Ωcalcite, is 0.87), the bottom water is corrosive to calcite. We investigated the potential influence of dissolution of the G. ruber and T. sacculifer foraminifera shells on the δ18O and δ11B values, and thus on the mean calcification depth estimates and pCO2 reconstructions.
For G. ruber, both δ18O and δ11B of the shell is insensitive to dissolution14,31,32,33 (Supplementary Text). This is also supported by the fact that the δ18O variation of G. ruber in core KR05-15 PC01 carries an identical environmental signal to those collected from a nearby sediment core that has an excellent preservation of foraminifera shells (MD97-2138; Fig. 1, Supplementary Fig. S5)29. For T. sacculifer, on the other hand, shell dissolution effects on its geochemistry is not as straightforward as G. ruber, which is because of a presence of the sac-like final chamber. The sac-like final chamber and a gametogenic calcite coating are precipitated during the last life stage of this animal30,33,34,35,36,37. During the reproduction, they migrate into the thermocline depth and release gametes, which is governed by lunar cycles30,34,35,37. The mean calcification depth of T. sacculifer in the WEP is deeper than global mean, likely due to thick mixed layer and chlorophyll maximum in the thermocline depth30. For example, Rebotim et al.37 estimate using plankton tow that the mean calcification depth of T. sacculifer is shallower in the subtropical North Atlantic, ~60 m. Furthermore, the sac is likely more resistant to corrosive bottom water, thus the measurement of T. sacculifer with sac leads to a lower temperature (lower δ18O) and lower pH (lower δ11B) reconstruction33,38. However, two observation support that the presence of the sac-like final chamber does not alter either the mean calcification depth estimate of T. sacculifer or the pCO2 estimate of upper-thermocline seawater in the WEP (see Supplementary Text, for more thorough discussion). First, T. sacculifer with a sac-like final chamber in core KR05-15 PC01 carries an identical environmental signal (that is Mg/Ca and δ18O) to those without sac collected from a nearby sediment core that has an excellent preservation of foraminifera shells (MD97-2138; Fig. 1, Supplementary Fig. S6)29. Second, δ11B values of G. sacculifer from core KR05-15 PC01 are consistent with those from core-top material from the Ontong Java Plateau which is not affected by dissolution14 (Fig. 1, Supplementary Fig. S7). Thus we conclude that relatively deeper calcification depth is due to location-specific habitat depth preferences of T. sacculifer, not due to dissolution effects. The reason of this is likely that the modest sedimentation rate of KR05-15 PC01 (4.5 cm/ky) enabled the fine preservation of foraminifera shells, even though the bottom water was undersaturated. It is also noteworthy that large T. sacculifer individuals are less influenced by dissolution, because of a relative proportion of sac versus other chambers are smaller in large individuals38. It is likely that dissolution effects is negligible through the record, because there are little variation in saturation state in the Pacific deep-water in glacial-interglacial timescales39. We also found that the selection of different Mg/Ca–water temperature relationships and consideration of an effect of partial dissolution of G. ruber shell28,31,40 has a negligible effect on the calculation of surface seawater pCO2 (Supplementary Text, Supplementary Fig. S8). Similarly we found that pH effect on Mg/Ca–water temperature relationships and different total alkalinity estimation scenario since the last glacial period has a negligible effect on the calculation of seawater pCO2 (Supplementary Text, Supplementary Figs S9–S11).
Surface Seawater pCO2
Today (at the reference year 2000 CE), the surface seawater pCO2 of the WEP has a similar value (360 μatm) to the atmospheric value (370 μatm) (Fig. 1)41,42. A CO2 neutral condition (seawater pCO2 is close to atmospheric pCO2) is also likely to have existed in pre-industrial times when the atmospheric pCO2 value was 280 μatm (ref.3). The pCO2 value of the surface seawater is calculated as 290 μatm (10 μatm higher than the atmospheric value) based on recent instrumental seawater CO2 system observations made using research vessels and theoretical calculations considering anthropogenic CO2 incorporation since pre-industrial times43 (Figs 2c,d and 3c). From the last glacial period to the early Holocene, reconstructed seawater pCO2 values of mixed-layer water obtained using G. ruber show variation similar to that of atmospheric values, suggesting that the oceanic surface layer in the WEP remained in a CO2 neutral condition (Fig. 2c,d). During the mid- Holocene (6.4–8.6 ka), the WEP became a modest source of CO2 (on average +36 μatm higher than the atmospheric pCO2), which is consistent with modern oceanographic observations (Fig. 2b,c), suggesting that G. ruber shells are a reliable recorder of surface seawater CO2 chemistry. Considering the estimation error of ΔpCO2 (the difference between seawater pCO2 and atmospheric pCO2) from the δ11B value of G. ruber, ΔpCO2 was near zero through the last deglaciation (Fig. 2d), showing that there is no evidence of CO2 release from the WEP during this period. This result is in clear contrast with a previous finding by Palmer and Pearson12 that the WEP was a CO2 source region. The reason for this is most likely that T. sacculifer (the species used by Palmer and Pearson12) record pH and pCO2 values of the upper thermocline, not of the surface water in the WEP. This point is also of great importance for the reconstruction of atmospheric CO2 using δ11B analysis of T. sacculifer shells for the deep past, beyond the limit of Antarctic ice cores (approximately, 800 ka)9,15,16,24, as a deeper calcification depth than the one we assume may result in the overestimation of atmospheric pCO2. We note that previous reconstruction using δ11B of T. sacculifer in the marine sediment core collected from the equatorial Atlantic is irrelevant to this issue (see the recent compilation by Dyez et al.15), because their calcification depth is confirmed to lie within mixed layer.
A Linkage Between High and Low Latitude Ocean
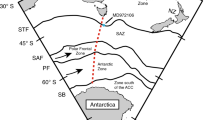
During the last glacial period, an efficient biological pump due to iron fertilization and increased stratification of the Southern Ocean due to sea ice expansion around Antarctica resulted in more efficient carbon storage in the deep sea and a drop in the atmospheric pCO2 level7,8,44 (Fig. 4). During the last deglaciation, upwelling in the Southern Ocean was enhanced, and carbon was returned to the atmosphere8,45,46. This event was likely accompanied by a stronger connection between the Southern Ocean and surface and thermocline depth of the low-latitude Pacific, which is known as “geochemical tunneling” (Fig. 4). A large variety of geochemical tracers have suggested greater enhancement of water transport due to Ekman pumping by the southward migration of southern westerlies7, more aged water mass incorporation into the intermediate water6,11,45,46,47, and more pronounced primary productivity in the Southern Ocean8,48. Higher surface pCO2 conditions during the last deglaciation reported for the EEP10 and the CEP (Tahiti)11,23 (Fig. 3) were likely a result of these phenomena, as such a water mass was likely accompanied by higher pCO2 water originating from the glacial deep-sea carbon reservoir (Fig. 4). The fact that no high-pCO2 water was observed in the mixed layer of the WEP suggests that the high-pCO2 region expanded southward but not westward (Fig. 4). Our results suggest that throughout deglaciation all of the nutrients/CO2 upwelled in the EEP were fully utilized by the time these surface waters arrived in the WEP. Additionally, the abovementioned 3-ky precedence of the increase in upper thermocline pCO2 (that is decrease in δ11B) in the WEP at the beginning of the last deglaciation (recorded in T. sacculifer shells) relative to the increase in surface pCO2 (recorded in G. ruber shells) was likely a result of an early arrival of high pCO2 water to the upper thermocline at approximately 19 ka. A possible source of such water mass is the Southern Ocean, especially via water transport via the Subantarctic Mode Water (SAMW) and Equatorial Undercurrent (Fig. 4). Considering that atmospheric CO2 began to increase only after about 17.5 ka, as determined from Antarctic ice core records (Fig. 2d), a stronger upwelling condition in the Southern Ocean and subsequent incorporation of high-pCO2 water into the SAMW might have started at 19 ka, although it was not enough to fuel atmospheric CO2. This idea is partly supported by the EEP surface ocean records at 19 ka that show significantly higher pCO2 than in the atmosphere (i.e., positive ΔpCO2)10 (Fig. 3). Another important observation is that surface ocean pCO2 in the EEP and the CEP and subsurface pCO2 in the WEP was remained high although atmospheric pCO2 did not increase during the Bølling–Allerød warm interval (B/A; 14.6–12.9 ka). Surface pCO2 reconstruction using δ11B of planktonic foraminifera, Neogloboquadrina pachyderma, collected from marine sediment core in the North Pacific (51.27°N, 167.73°E)49 shows similar variation to our subsurface pCO2 record, which may provide a hint to understand the mechanism behind, because there is another path that transport nutrient and carbon from the North Pacific to the equatorial Pacific via North Pacific Intermediate Water (NPIW)50. It is noteworthy that at the onset of the B/A interval, a collapse of NPIW formation is suggested49, which occurred at the same time of a reduction of the southern-sourced water contribution in the thermocline depth of the equatorial Pacific5. A hypothesis that pCO2 variability of thermocline in the equatorial Pacific is driven by both north and south high-latitude ocean processes merits testing by obtaining more high-resolution records from wider spatial coverage. In addition, other important research topics include investigating lower-thermocline conditions by utilizing thermocline-dwelling foraminifera shells (e.g., refs5,28) and investigating seawater pCO2 conditions during other glacial terminations, such as the penultimate deglaciation. For these purposes, the boron isotope proxy is a promising tool to create a pCO2 map in order to better understand the redistribution of CO2 between seawater and the atmosphere during glacial–interglacial cycles. Given a large surface area and its connectivity to the deep ocean, the Southern Ocean most likely had a central role in deglacial CO2 release into the atmosphere, but relative importance of CO2 flux from the Southern Ocean and that from other oceanic regions including the equatorial Pacific remains unquantified.

Schematics of deglacial marine carbon cycles. Schematics of carbon cycles and climatology in the South Pacific Ocean for three time periods: the last glacial period, the last deglaciation, and the Holocene.
Materials and Methods
Geochemical analyses
We analyzed a marine sediment core obtained from the West Caroline Basin (0.10°S 139.58°E) in 2005 by R/V Kairei. A 15-m-long sediment core that covers the last 400 ka was obtained using a piston coring system (core KR05-15 PC01)26. The top 1.5 m of the core was used for this study, as it covers the time period since the last glacial period. Sediment lithologies are foraminifera-rich clay (brown) for the top 30 cm and clay with foraminifera (greenish gray) from a depth of 30 cm to 150 cm. The archive half was cut in 2 cm intervals, and the sediment samples were gently washed under running water over a 63 μm sieve and dried at 60 °C in an oven. The sieved materials were further divided into size fractions of 63–255, 255–300, 300–355, 355–425, 425–500, and 500–850 μm, and stored in plastic vials.
The age model of the sediment core was established by 17 radiocarbon dates measured by accelerator mass spectrometry (AMS). If enough calcium carbonate materials were available, shells of mono-specific planktonic foraminifera (T. sacculifer) were used; otherwise, shells of mixed planktonic foraminifera were used (Supplementary Table S1). Seven samples were submitted as carbonate to the National Ocean Sciences Accelerator Mass Spectrometry Facility (NOSAMS), Woods Hole Oceanographic Institution (WHOI), and the remaining samples were analyzed in the Atmosphere and Ocean Research Institute (AORI), University of Tokyo. Three of the 20 samples were omitted as they showed anomalous values compared with neighboring data. Conventional 14C ages were converted into calendar age before 1950 CE using a Marine13 calibration curve51 with a local marine 14C reservoir age of 40 ± 21 years52,53 (Supplementary Fig. S1).
More than two shells of Uvigerina spp. were hand-picked under a microscope from the sediment samples from size fractions of 300–355 and 355–425 μm. Shells of G. ruber (white, s.s./s.l.) and T. sacculifer (with a sac-like final chamber) were hand-picked under microscope from size fractions of 300–355 and 500–850 μm, respectively. All samples were weighed, and if the sample amount was insufficient for the subsequent geochemical analysis, some continuous samples were mixed (at most eight samples).
Oxygen isotopes of benthic foraminifera Uvigerina spp. were measured using an isotope-ratio mass spectrometer (Isoprime) with an automated carbonate reaction system (Multiprep) installed at Kochi Core Center (KCC), Japan. The values of δ18O are reported with respect to the Vienna Pee Dee Belemnite standard using standard delta notation in permil54. Analytical precision for long-term measurement of the calcite standard (IAEA603) was better than 0.1‰. A drift correction for the sample δ18O values was performed, as the long-tem drift of the measured IAEA603 values of the standards during the measurement period between June 2017 and June 2018 was observed to be 0.12–0.32‰ isotopically heavier than the certified value of −2.37‰ (IAEA Reference Products; available at http://nucleus.iaea.org/rpst/ReferenceProducts/ReferenceMaterials/Stable_Isotopes/index.htm).
A technique for measuring Mg/Ca, δ18O, and δ11B from the same planktonic foraminifera sample was established in this study (Supplementary Fig. S2). The foraminifera cleaning methodology was after Baker et al.55. Weighed samples were crushed gently between two acrylic slides until the insides of all the chambers were visible under stereoscopic microscope. Samples were subsequently transferred to 1.5 mL Eppendorf tubes. Samples were sonicated five times with Milli-Q water and twice with methanol. The supernatant containing suspended fine grains was removed immediately from the sample after ultrasonication. To remove organic matter by oxidation, 0.1 N NaOH buffered 30% H2O2 was added and heated at 80 °C for 10 min. Subsequent reductive steps were skipped, as they do not have a discernible effect on δ11B measurement33,56.
Several hundred micrograms of samples were divided for δ18O measurements using the Isoprime spectrometer (Supplementary Fig. S2). The methodology of δ18O measurements was the same as above. The remaining samples were transferred to acid-cleaned Teflon vials. After weak acid-leaching of any adhesive materials attached to calcium carbonate, 1–2 mL 0.1 M HCl was added to the samples until complete dissolution, together with 5 μL of 1% mannitol solution. A total of 30 μL of sample was taken from the solution and diluted with 5 mL of 0.15 M HNO3 with internal standards (50 ppb Be and 100 ppb Sc, Y, and In). First, Ca concentration was measured using a quadrupole inductively coupled plasma mass spectrometer (Q-ICPMS; Agilent7700, Agilent Technology) installed at KCC (Supplementary Fig. S2). Then, 30 μL of sample was taken again from the solution and diluted to make the Ca concentration of each sample 10 ppm (matrix matching). Matrix matching is essential for the precise determination of trace element/Ca ratios28,57. Standard solution (200 ppb of Mg and Sr, 50 ppb of B, Al, Mn, Rb, and Ba) was prepared by diluting commercially available 1000 μg/g single element standards with the abovementioned 0.15 M HNO3 containing internal standards. Elemental ratios of samples were determined using the Agilent7700 instrument, with a machine drift correction. A calcium carbonate standard JCp-1, provided by the Geological Survey of Japan, was repeatedly analyzed to calculate the precision of the trace element/Ca ratio measurements. The precision of Mg/Ca analysis was 3.4% (1σ).
Boron isotopic measurements were conducted following the methodology of Tanimizu et al.58. Briefly, the boron was purified using cation- and anion-exchange resin columns, and the samples were dissolved with an acid mixture composed of 0.15 M HNO3, 0.05 M HF, and 0.1% mannitol to obtain a solution of 20–50 ppb B. Li standard (7Li/6Li isotopic reference) was added to each sample to correct a mass discrimination. δ11B values were determined with a multi-collector inductively coupled plasma mass spectrometer (MC-ICPMS) (Neptune, Thermo Finnigan) installed at KCC, against the isotopic reference NIST-SRM 951, using a standard-sample bracketing technique under wet plasma conditions. Since July 2017, a jet sampling cone instead of a normal sampling cone has been used with an X skimmer cone, which significantly increased beam intensity for both carbonate standard JCp-1 and seawater standard AE122 (Supplementary Fig. S3). δ11B values of JCp-1 were determined with different boron concentrations (10, 20, 30, 40, 50, and 75 ppb), which yielded consistent values with a mean of 24.38 ± 0.28‰ (2σ, n = 37) only if 11B beam intensity was greater than 0.2 V (Supplementary Fig. S3); in the case of beam intensity of less than 0.2 V, analytical error increased to ±0.5‰ (2σ, n = 10). δ11B values of AE122 were determined with a uniform concentration of 75 ppb, which yielded a mean value of 39.48 ± 0.24‰ (2σ, n = 14). As with JCp-1, the measurement of AE122 using a jet sampling cone resulted in higher beam intensity than measurement using a normal cone, and there were no discernible differences in δ11B between selections of sampling cones (Supplementary Fig. S3). δ11B values of both JCp-1 and AE122 were within the previously published 2σ range of data compiled from different laboratories by Foster et al.18 and Foster et al.59. The analytical error of JCp-1 (±0.14‰, 1σ) was taken as the analytical errors of the foraminifera samples, except when 11B beam intensity was less than 0.2 V, in which case the larger error (±0.25‰, 1σ) was employed.
To assess the reproducibility of the methodology, Mg/Ca, δ18O, and δ11B of T. sacculifer samples in the Holocene section were measured 2–4 times, as they were abundant in this interval. The results of the Mg/Ca, δ18O, and δ11B replicates showed good agreement within the estimated analytical errors (Supplementary Fig. S4), supporting the reproducibility of this methodology.
Age model
To obtain age-depth model of the sediment core KR05-15 PC01 in 1 cm intervals from 17 radiocarbon dates we used a recently published age-depth modeling tool “Undatable” (ref.27), which uses a deterministic approach with a positive sediment accumulation rate (Supplementary Fig. S1). We confirmed the robustness of the age model by comparing the δ18O values of Uvigerina spp. from core KR05-15 PC01 with a benthic δ18O record for Pacific deep-sea sediment60 (Supplementary Fig. S1). The values are in good agreement in the glacial period (30–18 ka), but there is an offset thereafter (maximum difference of 0.7‰). Although the absolute δ18O values are different, the shapes of the “up-down” patterns during 10–7 ka show excellent agreement. We infer that the difference is likely due to a regional difference in bottom-water temperature.
Data screening
Some δ11B data from planktonic foraminifera that showed anomalously low values (1–2‰) were discarded from the calculation of pCO2. Such data were accompanied by higher Mg/Ca ratios (by more than 0.3 mmol/mol) as well as high Al counts (typically more than 50,000 cps), suggesting that boron was contaminated by silicate in the sediment55,61. Barker et al.55 investigated how the Mg/Ca ratio of foraminifera shells changes according to physical and chemical cleaning steps, and clearly showed that high Mg/Ca ratios result from insufficient removal of silicate (which is a major component of marine sediment). Similarly, without cleaning, the Al/Ca ratio varies in concert with the Mg/Ca ratio of foraminifera55, suggesting that Mg/Ca and Al content are useful to identify contaminated samples. As foraminifera shells were dissolved in 0.1 M HCl, excess boron likely originated from some material that is reactive with HCl. Ishikawa and Nakamura61 reported that the δ11B value of the HCl-soluble fraction of subtropical North Pacific marine sediment (nanno oose; DSDP313 1–5; 20°N 170°W) was 8.0‰ with a significant boron abundance (15 ppm), which is much lower than δ11B values of G. ruber (20–22‰) and T. sacculifer (19–21‰). Thus, we infer that the anomalously low foraminiferal δ11B values observed in this study can be explained by contamination by boron that was not removed sufficiently during the cleaning steps.
Calculation of pH and pCO2
Water temperature and salinity need to be known for the calculation of seawater pH from the measured δ11B of foraminifera shells. Mg/Ca ratios of foraminifera shells were used for the calculation. The Mg/Ca–T equation of Sagawa et al.28 was used, as this equation was established by using a marine sediment core collected from a similar depth and location in the WEP (3cBX, 8.02°N, 139.64°E, 2829 m, Fig. 1).
where T is temperature in degrees Celsius.
Seawater δ18O (δ18OSW) was used to calculate salinity, as there is a linear relationship between salinity and δ18OSW. The δ18O of foraminifera calcite is a function of temperature and δ18OSW (ref.62), so δ18OSW can be calculated from δ18O and temperature (from Mg/Ca).
or
Here, α is the fractionation factor and T is temperature in degrees Celsius (after ref.63). However, in glacial–interglacial timescales, past δ18OSW changes need to be further considered for the calculation. Since the last glacial period, enormous volumes of freshwater with isotopically lighter δ18O values previously stored in continental ice sheets have entered the ocean, making global seawater δ18O lighter by ~1‰ (ice volume effect); as an additional consequence, sea level rose by ~135 m. As ice-derived δ18OSW change has a linear relationship with global sea level, we estimated the ice volume effect from the reconstructed sea level curve of Yokoyama et al.64. We assume that a sea level change of 135 m causes a 1‰ change in δ18OSW (ref.65). The ice volume-corrected δ18OSW values were subsequently used for the calculation of salinity using the reported equation of Schmidt66:
where S is salinity. There are known offsets between the theoretically expected δ11B of borate ions in the seawater (δ11Bborate) and the measured δ11B of foraminifera calcite (δ11Bborate), and these relationships differ among species (e.g., ref.13). For the calculation of seawater pH, the empirical relationships of Marinez-Boti et al.67 and Henehan et al.23 were used for G. ruber and T. sacculifer, respectively. Finally, seawater pH was obtained by the following theoretical equation:
where pKB is the dissociation constant of boric acid and is a function of temperature and salinity (and pressure)68, δ11BSW is the δ11B value of seawater, which is constant on glacial–interglacial timescales59, and α is the fractionation factor between borate ions and boric acid in the seawater69,70.
For the calculation of seawater pCO2, both pH and one more CO2 system parameter need to be known. In this study, the total alkalinity (TA) of the seawater was used for the calculation, and was estimated using a multiple regression equation (a function of temperature and salinity) reported by Lee et al.71. The CO2calc software (version 4.0.9), provided by the U.S. Geological Survey (available at https://www.usgs.gov/software/co2calc), was used to calculate pCO2 from pH, TA, temperature, and salinity. Dissociation constants for K1 and K2 from Lueker et al.72 and for KB from Dickson68 were used. ΔpCO2 (seawater pCO2 minus atmospheric pCO2) was calculated from the obtained seawater pCO2 using contemporary atmospheric pCO2 reconstructed from Antarctic ice cores1,2,3.
Estimated calcification depth of planktonic foraminifera
Calcification depths of G. ruber and T. sacculifer were estimated by comparing theoretically predicted depth profiles of δ18O and δ11B and measured δ18O and δ11B values in the Holocene section. For the calculation, depth profiles of annual temperature and salinity from the World Ocean Atlas (data available at https://odv.awi.de/data/ocean/world-ocean-atlas-2009/) were used (Fig. 3a). For CO2 system variables, gridded data of dissolved inorganic carbon (DIC) and TA from Global Ocean Data Analysis Project (GLODAP; data available at https://odv.awi.de/data/ocean/glodap-gridded-data/)43 were used. Anthropogenic DIC incorporation after the industrial revolution was subtracted from DIC data to estimate pre-industrial DIC, and then all CO2 system variables in the pre-industrial era were calculated using the CO2calc software (Fig. 3b,c). The theoretical δ18O depth profile of calcite was calculated from temperature and δ18OSW values obtained from NASA GISS (data available at http://iridl.ldeo.columbia.edu/SOURCES/.NASA/.GISS/.LeGrande_Schmidt2006/.v1p1/). As foraminifera calcite is precipitated in isotopic equilibrium with the ambient seawater, the δ18O depth profile of inorganic calcite represents that of foraminifera calcite (Fig. 3d). The δ11B depth profile of foraminifera calcite can be predicted from the known temperature, salinity, and pH, as well as an empirical relationship between the δ11B of seawater borate ion and that of G. ruber67 and T. sacculifer13 (Fig. 3e). As the empirical relationship differs among foraminifera species, depth profiles of the predicted δ11B of foraminifera shells differ between G. ruber and T. sacculifer (Fig. 3e). The estimated calcification depths are 0–75 m (within the mixed layer) and 75–125 m (below the mixed layer; upper thermocline) for G. ruber and T. sacculifer, respectively.
Monte Carlo simulation
As many variables are required to calculate pH, pCO2, and ΔpCO2, Monte Carlo simulation is useful to calculate the estimation errors of these outputs considering all involved errors. All calculations were performed using the freeware R (version 3.0; available at: https://www.r-project.org) with the seacarb (v.3.2.8) package for seawater CO2 chemistry calculation73. The same dissociation constants (K1, K2, and KB) that were used for the seawater CO2 chemistry calculation using the CO2calc software were employed. Analytical errors (1σ) of Mg/Ca (3%), δ18O (0.1‰), and δ11B (0.14‰ and 0.25‰ when 11B beam intensity was greater than 0.2 V and less than 0.2 V, respectively) were considered. Estimation errors of atmospheric pCO2 (2 μatm) reconstructed from Antarctic ice core were also considered. Variables were randomly changed in which errors are normally distributed (mean ± error), and the calculation was repeated 1000 times. Standard deviations of 1000 outputs of pH, pCO2, and ΔpCO2 were then calculated, and were regarded as estimation errors (Fig. 2b–d). The error ranged from 0.014 to 0.030 for pH, and from 9 to 32 μatm for both pCO2 and ΔpCO2.
References
Marcott, S. A. et al. Centennial-scale changes in the global carbon cycle during the last deglaciation. Nature 514, 616–619 (2014).
Ahn, J. & Brook, E. J. Siple Dome ice reveals two modes of millennial CO2 change during the last ice age. Nat. Commun., https://doi.org/10.1038/ncomms4723 (2014).
Lüthi, D. et al. High-resolution carbon dioxide concentration record 650,000–800,000 years before present. Nature 453, 379–382 (2008).
Basak, C. et al. Breakup of last glacial deep stratification in the South. Pacific. Science 359, 900–904 (2018).
Pena, L. D. et al. Rapid changes in meridional advection of Southern Ocean intermediate waters to the tropical Pacific during the last 30 kyr. Earth Planet. Sci. Let. 368, 20–32 (2013).
Skinner, L. C., Fallon, S., Waelbroeck, C., Michel, E. & Barker, S. Ventilation of the Deep Southern Ocean and Deglacial CO2 Rise. Science 328, 1147–1151 (2010).
Sigman, D. M., Hain, M. P. & Haug, G. H. The polar ocean and glacial cycles in atmospheric CO2 concentration. Nature 466, 47–55 (2010).
Anderson, R. F. et al. Wind-Driven Upwelling in the Southern Ocean and the Deglacial Rise in Atmospheric CO2. Science 323, 1443–1448 (2009).
Foster, G. L. & Rae, J. W. B. Reconstructing Ocean pH with Boron Isotopes in Foraminifera. Annu. Rev. Earth Planet. Sci. 44, 207–37 (2016).
Martinez-Boti, M. A. et al. Boron isotope evidence for oceanic carbon dioxide leakage during the last deglaciation. Nature 518, 219–222 (2015).
Kubota, K., Yokoyama, Y., Ishikawa, T., Obrochta, S. & Suzuki, A. Larger CO2 source at the equatorial Pacific during the last deglaciation. Sci. Rep., https://doi.org/10.1038/srep05261 (2014).
Palmer, M. R. & Pearson, P. N. A 23,000-Year Record of Surface Water pH and pCO2 in the Western Equatorial Pacific. Ocean. Science 300, 480–482 (2003).
Henehan, M. J. et al. A new boron isotope-pH calibration for Orbulina universa, with implications for understanding and accounting for ‘vital effects’. Earth Planet. Sci. Let. 454, 282–292 (2016).
Foster, G. L. Seawater pH, pCO2 and [CO3 2−] variations in the Caribbean Sea over the last 130 kyr: A boron isotope and B/Ca study of planktic foraminifera. Earth Planet. Sci. Let. 271, 254–266 (2008).
Dyez, K. A., Hönisch, B. & Schmidt, G. A. Early Pleistocene obliquity-scale pCO2 variability at ~1.5 million years ago. Paleoceanogr. Paleoclimatol. 33, 1270–1291 (2018).
Hemming, N. G. & Hönisch, B. Boron isotopes in marine carbonate sediments and the pH of the ocean. Dev. Mar. Geol. 1, 717–34 (2007).
Ni, Y., Foster, G. L. & Elliott, T. The accuracy of δ11B measurements of foraminifers. Chem. Geol. 274, 187–195 (2010).
Foster, G. L. et al. Interlaboratory comparison of boron isotope analyses of boric acid, seawater and marine CaCO3 by MC-ICPMS and NTIMS. Chem. Geol. 358, 1–14 (2013).
Farmer, J. R., Hönisch, B. & Uchikawa, J. Single laboratory comparison of MC-ICP-MS and N-TIMS boron isotope analyses in marine carbonates. Chem. Geol. 447, 173–182 (2016).
Vengosh, A., Chivas, A. R. & McCulloch, M. T. Direct determination of boron and chlorine isotopic compositions in geological materials by negative thermal-ionization mass spectrometry. Chem. Geol. 79, 333–343 (1989).
Lecuyer, C., Grandjean, P., Reynard, B., Albarede, F. & Telouk, P. 11B/10B analysis of geological materials by ICP-MS Plasma 54: application to the boron fractionation between brachiopod calcite and seawater. Chem. Geol. 186, 45–55 (2002).
Aggarwal, J. K., Sheppard, D., Mezger, K. & Pernicka, E. Precise and accurate determination of boron isotope ratios by multiple collector ICP-MS: origin of boron in the Ngawha geothermal system, New Zealand. Chem. Geol. 199, 31–42 (2003).
Douville, E. et al. Abrupt sea surface pH change at the end of the Younger Dryas in the central sub-equatorial Pacific inferred from boron isotope abundance in corals (Porites). Biogeosciences 7, 2445–2459 (2010).
Chalk, T. B. et al. Causes of ice age intensification across the mid-Pleistocene transition. Proc. Natl Acad. Sci. USA 114, 13114–13119 (2017).
Picaut, J., Ioualalen, M., Menkes, C., Delcroix, T. & McPhaden, M. J. Mechanism of the Zonal displacements of the Pacific warm pool: implications for ENSO. Science 274, 1486–1489 (1996).
Yamazaki, T., Kanamatsu, T., Mizuno, S., Hokanishi, N. & Gaffar, E. Z. Geomagnetic field variations during the last 400 kyr in the western equatorial Pacific: Paleointensity-inclination correlation revisited. Geophys. Res. Let. 35, L20307, https://doi.org/10.1029/2008GL035373 (2008).
Lougheed, B. C. & Obrochta, S. P. A rapid, deterministic age-depth modeling routine for geological sequences with inherent depth uncertainty. Paleoceanogr. Paleoclimatol. 34, 122–133 (2019).
Sagawa, T., Yokoyama, Y., Ikehara, M. & Kuwae, M. Shoaling of the western equatorial Pacific thermocline during the last glacial maximum inferred from multispecies temperature reconstruction of planktonic foraminifera. Palaeogeogr. Palaeoclim. Palaeoecol. 346–347, 120–129 (2012).
de Garidel-Thoron, T. et al. A multiproxy assessment of the western equatorial Pacific hydrography during the last 30 kyr. Paleoceanogr. 22, PA3204, https://doi.org/10.1029/2006PA001269 (2007).
Rippert, N. et al. Constraining foraminiferal calcification depths in the western Pacific warm pool. Mar. Micropaleontol. 128, 14–27 (2016).
Dekens, P. S., Lea, D. W., Pak, D. K. & Spero, H. J. Core top calibration of Mg/Ca in tropical foraminifera: Refining paleotemperature estimation. Geochem. Geophys. Geosys. 3, https://doi.org/10.1029/2001GC000200 (2002).
Fehrenbacher, J. & Martin, P. Western equatorial Pacific deep water carbonate chemistry during the Last Glacial Maximum and deglaciation: Using planktic foraminiferal Mg/Ca to reconstruct sea surface temperature and seafloor dissolution. Paleoceanography 26, PA2225, https://doi.org/10.1029/2010PA002035 (2011).
Ni, Y. et al. A core top assessment of proxies for the ocean carbonate system in surface-dwelling foraminifers. Paleoceanography 22, PA3212, https://doi.org/10.1029/2006PA001337 (2007).
Bijma, J. & Hemleben, C. Population dynamics of the planktic foraminifer Globigerinoides sacculifer (Brady) from the central Red Sea. Deep-Sea Res., Part 1, Oceanogr. Res. Pap. 41, 485–510 (1994).
Erez, J., Almogi-Labin, A. & Avraham, S. On the life history of planktonic foraminifera: lunar reproduction cycle in Globigerinoides sacculifer (Brady). Paleoceanography 6, 295–306 (1991).
Be, A. W. H. Gametogenic calcification in a spinose planktonic foraminifer, Globigerinoides sacculifer (Brady). Mar. Micropaleontol. 5, 283–310 (1980).
Rebotim, A. et al. Factors controlling the depth habitat of planktonic foraminifera in the subtropical eastern North Atlantic. Biogeosciences 14, 827–859 (2017).
Hönisch, B. & Hemming, N. G. Ground-truthing the boron isotope-paleo-pH proxy in planktonic foraminifera shells: Partial dissolution and shell size effects. Paleoceanography 19, PA4010, https://doi.org/10.1029/2004PA001026 (2004).
Yu, J. et al. Loss of carbon from the deep sea since the Last Glacial Maximum. Science 330, 1084–1087 (2010).
Rosenthal, Y. & Lohmann, G. P. Accurate estimation of sea surface temperatures using dissolution-corrected calibrations for Mg/Ca paleothermometry. Paleoceanography 17, 1044, https://doi.org/10.1029/2001PA000749 (2002).
Midorikawa, T. et al. Y. Decreasing pH trend estimated from 25-yr time series of carbonate parameters in the western North Pacific. Tellus 62B, 649–659 (2010).
Takahashi, T. et al. Climatological mean and decadal change in surface ocean pCO2, and net sea–air CO2 flux over the global oceans. Deep-Sea Res. 56, 554–577 (2009).
Key, R. M. et al. A global ocean carbon climatology: Results from Global Data Analysis Project (GLODAP). Global Biogeochem. Cy., https://doi.org/10.1029/2004GB002247 (2004).
Jaccard, S. L., Galbraith, E. D., Martínez-García, A. & Anderson, R. F. Covariation of deep Southern Ocean oxygenation and atmospheric CO2 through the last ice age. Nature 530, 207–210 (2016).
Marchitto, T. M., Lehman, S. J., Ortiz, J. D., Flückiger, J. & van Geen, A. Marine Radiocarbon Evidence for the Mechanism of Deglacial Atmospheric CO2 Rise. Science 316, 1456–1459 (2007).
Ronge, T. A. et al. Radiocarbon constraints on the extent and evolution of the South Pacific glacial carbon pool. Nat. Commun. 7:11487, https://doi.org/10.1038/ncomms11487.
Basak, C., Martin, E. E., Horikawa, K. & Marchitto, T. M. Southern Ocean source of 14C-depleted carbon in the North Pacific Ocean during the last deglaciation. Nat. Geosci. 3, 770–773 (2010).
Horn, M. G., Beucher, C. P., Robinson, R. S. & Brzezinski, M. A. Southern ocean nitrogen and silicon dynamics during the last deglaciation. Earth Planet. Sci. Let. 310, 334–339 (2011).
Gray, W. R. et al. Deglacial upwelling, productivity and CO2 outgassing in the North Pacific Ocean. Nat. Geosci. 11, 340–344 (2018).
Sarmiento, J. Á., Gruber, N., Brzezinski, M. A. & Dunne, J. P. High-latitude controls of thermocline nutrients and low latitude biological productivity. Nature 427, 56–60 (2004).
Reimer, P. J. et al. IntCal13 and Marine13 radiocarbon age calibration curves 0–50,000 years cal BP. Radiocarbon 55, 1869–1887 (2013).
Petchey, F. & Ulm, S. Marine Reservoir Variation in the Bismarck Region: An Evaluation of Spatial and Temporal Change in ΔR and R Over the Last 3000 Years. Radiocarbon 54, 45–58 (2012).
McGregor, H. V., Gagan, M. K., McCulloch, M. T., Hodge, E. & Mortimer, G. Mid-Holocene variability in the marine 14C reservoir age for northern coastal Papua New Guinea. Quat. Geochron. 3, 213–225 (2008).
Craig, H. Isotopic standards for carbon and oxygen and correc- tion factors for mass-spectrometric analysis of carbon dioxide. Geochim. Cosmochim. Acta 12, 133–149 (1957).
Barker, S., Greaves, M. & Elderfield, H. A study of cleaning procedures used for foraminiferal Mg/Ca paleothermometry. Geochem. Geophys. Geosys. 4, https://doi.org/10.1029/2003GC000559 (2003).
Misra, S., Owen, R., Kerr, J., Greaves, M. & Elderfield, H. Determination of δ11B by HR- ICP-MS from mass limited samples: application to natural carbonates and water samples. Geochim. Cosmochim. Acta 140, 531–552 (2014).
de Villiers, S., Greaves, M. & Elderfield, H. An intensity ratio calibration method for the accurate determination of Mg/Ca and Sr/Ca of marine carbonates by ICP-AES. Geochem. Geophys. Geosys. 3, 1001, https://doi.org/10.1029/2001GC000169 (2002).
Tanimizu, M., Nagaishi, K. & Ishikawa, T. A Rapid and Precise Determination of Boron Isotope Ratio in Water and Carbonate Samples by Multiple Collector ICP-MS. Anal. Sci. 34, 667–674 (2018).
Foster, G. L., Pogge von Strandmann, P. A. E. & Rae, J. W. B. Boron and magnesium isotopic composition of seawater. Geochem. Geophys. Geosys. 11, Q08015 (2010).
Lisiecki, L. E. & Raymo, M. E. Diachronous benthic δ18O responses during late Pleistocene terminations. Paleoceanography 24, PA3210 (2009).
Ishikawa, T. & Nakamura, E. Boron isotope systematics of marine sediments. Earth Planet. Sci. Let. 117, 567–580 (1993).
Kim, S.-T. & O’Neil, J. R. Equilibrium and nonequilibrium oxygen isotope effects in synthetic carbonates. Geochim. Cosmochim. Acta 61, 3461–3475 (1997).
Bemis, B. E., Spero, H. J., Bijma, J. & Lea, D. W. Reevaluation of the oxygen isotopic composition of planktonic foraminifera: Experimental results and revised paleotemperature equations. Paleoceanography 13, 150–160 (1998).
Yokoyama, Y. et al. Rapid glaciation and a two-step sea level plunge into the Last Glacial Maximum. Nature 559, 603–607 (2018).
Adkins, J. F., McIntyre, K. & Schrag, D. P. The Salinity, Temperature, and δ18O of the Glacial Deep. Ocean. Science 298, 1769–1773 (2002).
Schmidt, G. A. Error analysis of paleo-salinity calculations. Paleoceanography 14, 422–429 (1999).
Martínez-Boti, M. A. et al. Plio-Pleistocene climate sensitivity evaluated using high-resolution CO2 records. Nature 518, 49–54 (2015).
Dickson, A. G. Thermodynamics of the dissociation of boric acid in synthetic seawater from 273.15 to 318.15 K. Deep-Sea Res. 37, 755–766 (1990).
Klochko, K., Kaufman, A. J., Yao, W., Byrne, R. H. & Tossell, J. A. Experimental measurement of boron isotope fractionation in seawater. Earth Planet. Sci. Let. 248, 276–285 (2006).
Nir, O., Vengosh, A., Harkness, J. S., Dwyer, G. S. & Lahav, O. Direct measurement of the boron isotope fractionation factor: reducing the uncertainty in reconstructing ocean paleo-pH. Earth Planet. Sci. Let. 414, 1–5 (2015).
Lee, K. et al. Global relationships of total alkalinity with salinity and temperature in surface waters of the world’s oceans. Geophys. Res. Let. 33, L19605, https://doi.org/10.1029/2006GL027207 (2006).
Lueker, T. J., Dickson, A. G. & Keeling, C. D. Ocean pCO2 calculated from dissolved inorganic carbon, alkalinity, and equations for K-1 and K-2: validation based on laboratory measurements of CO2 in gas and seawater at equilibrium. Mar. Chem. 70, 105–119 (2000).
Lavigne, H., Epitalon, J. M. & Gattuso J. P. seacarb: seawater carbonate chemistry with R. R package version 3.0, http://CRAN.R-project.org/package=seacarb (2011).
Locarnini, R. A. et al. World Ocean Atlas 2009, Volume 1: Temperature. S. Levitus, Ed. NOAA Atlas NESDIS 68, US. Government Printing Office, Washington, D.C., 184 pp. (2010).
Acknowledgements
We would like to thank N. Ahagon for core curation and assistance in sediment core sampling; H. Takagi for assistance in planktonic foraminifera picking; K. Nagaishi, T. Kawai, Y. Nagaiwa, and N. Asakura for laboratory management and technical support in Mg/Ca and δ11B analyses; Y. Fujimura and T. Komatsu for technical support in δ18O analysis; and Y. Miyairi, T. Aze, S. Izawa, and Y. Ando for technical support in 14C analyses. We acknowledge that marine sediment core was obtained from JAMSTEC Core Samples for the scientific purpose. This study was financially supported by Research Fellowships for Young Scientists from the Japan Society for the Promotion of Science (JSPS) to K.K. (Grant Number: 17J04576); JSPS Kakenhi grants to K.K. (Grant Number: 18K18186); to T.I. (Grant Number: 16H04066); and to T.S. (Grant Number: 15K21221); Grant for Environmental Research Projects from The Sumitomo Foundation to T.S. (Grant Number: 133303).
Author information
Authors and Affiliations
Contributions
T.Y. obtained the marine sediment core. K.K., Y.Y. and T.S. designed the study and obtained foraminifera samples. K.K. and T.I. measured the Mg/Ca and boron isotope ratios of foraminifera samples. K.K. and M.I. measured oxygen isotope ratios of foraminifera samples. K.K., Y.Y., T.I., T.S., M.I. and T.Y. contributed to the interpretation of the data and the preparation of the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kubota, K., Yokoyama, Y., Ishikawa, T. et al. Equatorial Pacific seawater pCO2 variability since the last glacial period. Sci Rep 9, 13814 (2019). https://doi.org/10.1038/s41598-019-49739-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49739-0
This article is cited by
-
Southern Ocean glacial conditions and their influence on deglacial events
Nature Reviews Earth & Environment (2023)
-
Plutonium isotopes in the North Western Pacific sediments coupled with radiocarbon in corals recording precise timing of the Anthropocene
Scientific Reports (2022)
-
Abrupt upwelling and CO2 outgassing episodes in the north-eastern Arabian Sea since mid-Holocene
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
