
Abstract
The large-scale practical application of fuel cells cannot come true if the high-priced Pt-based electrocatalysts for oxygen reduction reaction (ORR) cannot be replaced by other efficient, low-cost and stable electrodes. Here, based on density functional theory (DFT), we exploited the potentials of layered SiC sheets as a novel catalyst for ORR. From our DFT results, it can be predicted that layered SiC sheets exhibit excellent ORR catalytic activity without CO poisoning, while the CO poisoning is the major drawback in conventional Pt-based catalysts. Furthermore, the layered SiC sheets in alkaline media has better catalytic activity than Pt(111) surface and have potential as a metal-free catalyst for ORR in fuel cells.
Similar content being viewed by others
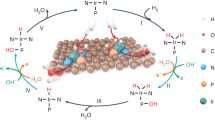


Introduction
As one of the most promising power sources, fuel cells have received considerable attention due to their high efficiency and low environmental impact. The sluggish kinetics of the oxygen reduction reaction (ORR) at the cathode is one of the key factors limiting the performance of fuel cells and efficient ORR electrocatalysts are essential for practical applications of the fuel cells. Pt has conventionally been employed as the cathode catalyst due to its high activity for the ORR1,2. The high cost, limited supply and poor durability of Pt catalyst however hinders the large-scale commercialization of fuel cells. Hence, during the last few decades, numerous studies have been devoted to find alternative electrocatalysts for the cathode side of fuel cells, including Pt-based alloys3,4,5,6, Pt-based core-shell/alloy nanoparticles7,8,9, carbon nanotubes-supported metal particles10,11, graphitic carbon nitride/carbon composite12,13 N/B/P/S doped carbon nanotubes (CNTs) and graphenes14,15,16.
Carbon-based catalysts are expected to be the most promising alternatives to Pt catalysts because C is more abundant and durable, as well as less expensive than Pt. The introduction of N (or B, P, S) atoms into sp2-hybridized carbon frameworks of graphenes or CNTs is generally effective in modifying their electrical properties and chemical activities17,18. Recent studies have confirmed that N, B, P and S doped carbon materials are hopeful candidates to replace Pt-based catalysts for fuel cells due to their high catalytic activity, long-term stability and excellent CO tolerance14,15,19,20,21,22,23. Theoretical studies have confirmed that the improved electrocatalytic activity of these materials can be attributed to the changes of electronic structure by doping N, B, P and S into carbon materials24,25. However, the dopant concentration in present carbon-based catalysts is low (N ~ 4–6 at%, B ~ 0–2.24 at% and 1–2 at% of S)19,20,21, which limits the improvement of the catalytic activity of carbon-based catalysts.
SiC has a unique combination of high saturated carrier mobility, high critical electrical field and high thermal conductivity. SiC with the atom ratio of 1:1 can provide more active reaction sites than present doped carbon materials to serve as a feasible metal-free ORR electrocatalyst. Herein, we predicted that layered SiC sheets exhibit excellent ORR catalytic activity and high CO tolerance and may be a very likely candidate for the next generation of low-cost ORR electrocatalysts.
Results
Firstly, the relative stability between layered and cubic SiC sheets, which is a function of layer number N, is determined by calculating the energy difference ΔE = Elayered − Ecubic, where Elayered and Ecubic are the total energies of layered and cubic SiC sheets, respectively. The layered SiC sheets are energetically favorable in comparison to cubic SiC sheets when N < 4, while cubic SiC sheets are advantaged as N ≥ 4, as shown in Fig. S1 and Table S1 in supporting information. Similar to graphite, layered SiC sheets usually show weak layer-layer interactions. Even further considering the van der Waals bonding, layered SiC sheets are still energetically more favorable than cubic SiC sheets when N < 4. This is consistent with the recent experimental results that the thickness of synthesized 2D SiC nanosheets is between 0.5 and 1.5 nm26.
We then turn to consider the adsorption and reduction of O2 on a single-layer SiC. The adsorption energies of adsorbates on layered SiC sheets are determined by Ead = Eads + ESiC − Eads/SiC, where Eads, ESiC and Eads/SiC are the total energies of the isolated adsorbate molecule, the layered SiC sheets and the adsorption systems, respectively. Positive adsorption energies correspond to exothermic adsorption processes, while negative adsorption energies do endothermic ones. Several possible high-symmetry adsorption sites are considered, including the top, bridge and hollow sites. The most stable adsorption structures and energies of O2 and ORR intermediates are summarized in Fig. 1 and Table 1. The meta-stable adsorption structures and energies are displayed in Fig. S2 of supporting information. It is found that the most energetically favorable site for O2 adsorption on the single-layer SiC is characterized by O2 nearly parallel to the SiC bond with adsorption energy of 0.56 eV. The O-O bond is elongated by 0.29 Å due to the electronic charge transfer from SiC to the 2π* orbital of O2, indicating that O2 can be dissociated easily on SiC. The charge transfer from the single-layer SiC to O2 molecule is quantified as 0.34 e according to Hirshfeld population charge distribution. Furthermore, the maximum coverage of O2 on SiC is also explicitly identified. As shown in Table S2 and Fig. S3 of supporting information, only two O2 molecules can be adsorbed on the single-layer SiC with 2 × 2 supercell, corresponding to a coverage 0.5 ML (monolayer) (1 ML is defined as one O atom per surface atom). The adsorption energy of the first O2 molecules on single-layer SiC is 0.48 eV. The second O2 molecule has two distinct adsorption structures with adsorption energies of 0.61 and 0.13 eV, respectively. It is found that the energetically more favorable configuration of second O2 forms two chemical bonds with Si atoms. This special adsorption structure results in the stronger adsorption of the second O2 molecule than the first one. This is because the electronegativity of Si (1.90) is smaller than that of C (2.55) while the Si-O bond is stronger than the C-O bond. Similar results are also found in 4 × 4 supercell, where the coverage also reaches 0.5 ML. Furthermore, O2 adsorbed as ordered structures in a 4 × 4 supercell is considered, as shown in Fig. S3 of supporting information, the corresponding coverage can reach 0.75 ML. However, the coverage of O2 on Pt(111) surface can reach 1 ML. All the O2 molecules adsorb on the bridge sites. This is consistent with previous theoretical work about O2 adsorption on Pt(111) surface27,28. Although the coverage of O2 on layered SiC is smaller than that on Pt(111) surface, the remaining adsorption sites on single-layer SiC is favorable for adsorption of other reactants, which may be beneficial for ORR.

Optimized adsorption structures for ORR intermediates on a single-layer SiC: (a) O2, (b) O, (c) OH, (d) H, (e) H2O and (f) CO.
Gray, gold, white and red colors denote C, Si, H and O atoms.
The most stable adsorption site for O atom is the bridge site, which gives rise to a highly stable epoxide-like structure with adsorption energy of 4.18 eV. In addition, H and OH prefer to adsorbing at atop site of Si atom other than that of C atom with the adsorption energies of 1.32 and 2.80 eV, respectively, which differs from the case in N-doped carbon materials29. Because H and OH tend to adsorb at positive charged adsorption sites. Note that H2O is stable on atop site of Si atom of the single-layer SiC. In contrast, H2O dissociates to H and OH spontaneously on cubic SiC sheets, which may prevent ORR on cubic SiC sheets. The reason is that Si and C atoms are sp3 hybridized in cubic SiC sheets, while they are sp2 in layered ones. The dangling bonds of Si or C atoms at the surface of cubic SiC sheets are very active. When H is introduced to O2, no matter whether the presence of H2O molecules around, O-O bond dissociates to form O and OH spontaneously, as shown in Fig. S4 of supporting information. OOH cannot exist as a stable intermediate in the ORR processes. And then, H2O2 cannot be produced based on OOH. Thus, OOH and H2O2 pathways of ORR on the single-layer SiC are neglected here30,31.
For CO adsorption, adsorption energy value changes from −0.08 eV to 0.07 eV after considering the effect of van der Waals bonding. Since the small adsorption energy value of CO adsorption, layered SiC sheets show excellent CO tolerance. In contrast, the adsorption energy of CO on Pt(111) surface (1.86 eV) is twice as large as that of O2 (0.84 eV), as shown in Table S3 of supporting information. Therefore, CO blocks the active sites and hinders the ORR on Pt(111) surface32,33. From partial density of states (PDOS) (Fig. S5 of supporting information), it is found that the hybridization between CO molecule and Si atom is very small, suggesting that the interaction between CO and SiC is little. By contrast, the interaction between CO and Pt(111) surface is very strong. All the main orbitals of CO hybridize with Pt states. The renowned mechanism of CO-metal interaction, namely, via donation from CO-5σ to metals and back-donation from metals to CO-2π* orbital, is applicable for CO adsorbed on Pt(111)34,35. This is confirmed by the fact of electron depletion at CO-5σ and partially occupied CO-2π*, which is empty above the Fermi level in gas CO. Consequently, CO will poison the Pt(111) surface.
It is known that ORR mechanisms at cathodes in acidic and alkaline solutions are different. In an acidic solution, the electrode reaction can be written as O2 + 4H+ + 4e− → 2O + 4H+ + 4e− → O + OH + 3H+ + 3e− → O + H2O + 2H+ + 2e− → OH + H2O + H+ + e− → 2H2O, while in an alkaline solution that can be expressed as O2 + 2H2O + 4e− → O + 2OH− + H2O + 2e− → 4OH−. Both reaction pathways are considered here. In general, ORR can proceed in Langmuir-Hinshelwood (LH) or Eley-Rideal (ER) mechanisms. LH mechanism involves all the reacting intermediates on the surface, whereas ER mechanism involves species from the electrolyte reacting with a surface intermediate. Both mechanisms are also taken into account one by one.
The ORR following the LH mechanism is discussed firstly. The ORR mechanism on the single-layer SiC in LH mechanism under acidic media can be divided into five elemental steps: O2 dissociation, two O atom hydrogenation steps and two OH hydrogenation steps, as shown in Fig. 2. Some possible ORR pathways with higher barrier energies are shown in Fig. S6 of supporting information. The activation energies for the five elemental steps are 0.29 eV for O2 dissociation, 0.43 and 0.49 eV for first and second OH formation, 1.11 and 1.05 eV for first and second H2O formation, respectively. The corresponding reaction energies are −1.61, −1.13, −1.00, 0.44 and 0.51 eV. H2O formation is the rate-determining step (RDS) in the O2 dissociation mechanism of ORR in the above process. The Si-H bond broken from the initial state to the transition state contributes to the most part of the activation energy of H2O formation. As the number of H atoms introduced to the O atom increases, the activation energy increases from 0.49 eV to 1.05 eV and the reaction energy increases from −1 eV to 0.51 eV, corresponding to the Brønsted-Evans-Polanyi relation between activation energy and reaction energy in the heterogeneous catalysis36,37,38. Activation energy is a almost linear function of reaction energy and reactions belong to the same class even follow the same relation36,37,38. The decrease of the activity of O atom is due to the increase of the number of introduced H atoms.

Minimum energy pathways for ORR elemental steps in acidic media on a single-layer SiC: (a) O2 → 2O, (b) 2O + H → O + OH, (c) O + OH + H → O + H2O, (d) O + H → OH, (e) OH + H → H2O and (f) schematic energy profile.
IS, TS and FS are initial, transition and final states, respectively. Gray, gold, white and red colors denote C, Si, H and O atoms.
ORR on the single-layer SiC in LH mechanism under an alkaline environment can be divided into the two elemental steps: O2 + H2O → O + 2OH and O + H2O → 2OH, as shown in Fig. 3. Differing from that in acidic environment, both steps are easy to cross with small activation energies (0.11 and 0.23 eV) and exothermic reaction energies (−2.20 and −0.40 eV). These can be deduced from the special geometrical parameters along reaction paths. O and H atoms only need a little movement from the initial state to the transition state, which costs little energy. Furthermore, the hydrogen bond between O atom and H atom in H2O molecule also reduce the energy barrier for H atom transfer. The activation energies for isolated H2O and O2 dissociations are 0.56 and 0.29 eV, both of them are surmountable at room temperature. This is consistent with the small activation energies for ORR elemental steps in alkaline media. Thus, we predict that ORR in LH mechanism on the single-layer SiC in the alkaline media is more favorable than that in the acidic media.

Minimum energy pathways for ORR elemental steps in alkaline media on a single-layer SiC: (a) O2 + H2O → O + 2OH, (b) O + H2O → 2OH and (c) schematic energy profile.
IS, TS, MS and FS are initial, transition, metastable and final states, respectively. ⋆ denotes the TS in each step. Gray, gold, white and red colors denote C, Si, H and O atoms.
As N increases from 1 to 3, adsorption energies for ORR intermediates, activation and reaction energies for ORR elemental steps in LH mechanism on layered SiC sheets are almost unchanged as shown in Tables 1 and 2. Thus, layered SiC sheets with different N values should show similar ORR catalytic activities.
For a comparison purpose, ORR elemental steps in LH mechanism on Pt(111) surface are also calculated, as depicted in Fig. S7 and Tables S4 of supporting information. The five barriers in O2 dissociation mechanism are calculated to be 0.85 eV for O2 dissociation, 1.00 and 1.22 eV for two OH formation steps and 0.38 eV for H2O formation. For the OOH association mechanism, it is found that the barriers are 0.61 eV for OOH formation, 0.03 eV for OOH dissociation, 1.22 eV for OH formation and 0.38 eV for H2O formation. OH formation is RDS for both the O2 dissociation and OOH association mechanisms. This is consistent with the recent literature30. The RDS of ORR on Pt(111) surface in acidic media is the O atom hydrogenation to OH with activation energy of 1.22 eV, which is similar to that of ORR on layered SiC sheets, suggesting that layered SiC sheets and Pt(111) surface may exhibit similar activities for ORR in LH mechanism under an acidic environment. In the alkaline media, however, the activation energies for the two elemental ORR steps in LH mechanism on Pt(111) surface are 0.43 and 0.55 eV, respectively, both of them are larger than that on layered SiC sheets since the bond length of Pt-Pt (2.77 Å) is longer than that of Si-C (1.79 Å). The energy cost for the longer distance diffusion of H atom on Pt(111) surface is larger than that on layered SiC sheets. In addition, the elemental step of O + H2O → 2OH, which is the RDS of ORR in LH mechanism on Pt(111) surface in alkaline media, is endothermic by 0.51 eV, while that on layered SiC sheets is exothermic with reaction energy of nearly −0.40 eV, suggesting that ORR in LH mechanism on layered SiC sheets is more advantaged than that on Pt(111) surface in alkaline environment.
Although energy diagram of ORR in ER mechanism is quite different from that in LH mechanism, both of which show similar results somehow. As shown in Fig. 4(a), ORR in ER mechanism exhibits exothermic reactivity at electrode potential U = 0 V under an acidic medium. As the pH value increases, the energy of each net coupled proton and electron transfer (CPET) step is shifted due to the effect of concentration on the free energy of H+. At pH = 1, the reaction energies of five elemental steps calculated are −1.61 eV for O2 dissociation, −1.44 and −1.32 eV for first and second OH formation, −0.06 and −0.34 eV for first and second H2O formation, respectively. When pH ≥ 4, the reaction energy of first H2O formation changes from exothermic to endothermic, while other steps remain exothermic. As U increases from 0 V to the ideal electrode potential of 1.23 V, energy levels are shifted for each net CPET step31,39. The reaction energies of the reactions in the LH mechanism and O2 dissociation in the ER mechanism remain unchanged. This is because no CPET step involves in these steps and the electrode potential only affects the energy levels of the electrons in the CPET steps31,39. At U = 1.23 V, OH formation in ER mechanism is still exothermic, while two H2O formation steps change to 1.17 and 0.89 eV endothermically at pH = 1, respectively. Consistent with ORR in LH mechanism, H2O formation is also the RDS of ORR on single SiC layer in ER mechanism at U = 1.23 V. The high concentration of H2O in electrolyte can improve the rate constant for the backward reaction and may have a negative effect on the entire process. Just as any coin has two sides, the issue being discussed here is no exception. H2O solution also has some positive effects on the ORR. As shown in Table S5 of supporting information, the adsorption of O2 is enhanced in H2O solution, resulting in the increase of reactant concentration on catalyst surface and forward reaction rate.

Schematic energy profile (relative to molecular O2 + 2H2 in acidic media and molecule O2 + 2H2O in alkaline media) for the ORR pathway on a single-layer SiC: (a) in acidic media, (b) in alkaline media.
A substantially different picture is obtained for ORR in ER mechanism under alkaline environment. Similar with that under acidic media, pH imports similar effect and shifts up the energy level of every CPET step. As shown in Fig. 4(b), at U = 0 V, adsorbed O2 molecule reaction with H2O is exothermic with reaction energy of −1.40 eV, while O atom reaction with H2O is endothermic with reaction energy of 0.06 eV at pH = 14. When an ideal electrode potential of 0.40 V is applied, the energy levels of every CPET step are shifted up by 0.80 eV for every double CPET step. The reaction energies of O2 molecule reaction with H2O and O atom reaction with H2O vary to −0.60 and 0.86 eV at pH = 14. O2 molecule reaction with H2O is the RDS of ORR in ER mechanism under alkaline environment and the energy barrier increases as electrode potential increases.
ORR in ER mechanism on Pt(111) surface is also calculated. As shown in Fig. S8(a) of supporting information, all elemental steps involved in ORR via ER mechanism on Pt(111) are exothermic at pH = 1 and U = 0 V, while OH, H2O and OOH formations are 0.69, 0.53 and 0.63 eV endothermic at U = 1.23 V, which are smaller than the barrier of the RDS step (H2O formation) on single-layer SiC. These results are consistent with that obtained by Nørskov et al, where the reaction energy of OH and H2O formations change form exothermic to endothermic at U = 1.23 V and the OOH associative mechanism has the lowest barrier and dominates at low oxygen coverges40,41. Fig. S8(b) of supporting information also shows the energy diagram of ORR in ER mechanism on Pt(111) under alkaline environment. At U = 0 V and pH = 1, the reaction energies of O2 molecule reaction with H2O and O atom reaction with H2O on Pt(111) surface are −1.42 and 0.38 eV, respectively. At U = 0.40 V and pH = 1, the reaction energies of these two elemental steps vary to −0.62 and 1.18 eV, respectively. O2 molecule reaction with O atom is the RDS and the barrier are larger than that on single SiC layer, suggesting that layered SiC sheets exhibit better catalytic activity than Pt(111) surface in ER mechanism under alkaline environment.
Based on Eyring's canonical transition state theory, our calculations can be incorporated into a reduced kinetic model that should report qualitative features of the reaction mechanisms at different applied potentials39,42,43. As displayed in Fig. 5(a), the RDS of ORR in ER mechanism is O2 dissociation at low potential region, while it changes to H2O formation when U > 0.63 V at pH = 1. As shown in Fig. 5(b), O2 reaction with H2O is the RDS of ORR in ER mechanism under alkaline environment and the reaction rate decreases as electrode potential increases. Comparing Fig. 5 with Fig. S9 of supporting information, we can predict that the catalytic activity of layered SiC sheets is better than that of Pt(111) surface in alkaline media. Furthermore, due to the higher doped ratio, SiC can provide more ORR active sites than present doped carbon materials and may act as a feasible low-cost metal-free ORR electrocatalyst.

Potential-dependent rate constants for ORR on a single-layer SiC: (a) in acidic media with pH = 1, (b) in alkaline media with pH = 14.
Discussion
Since O2 adsorption and dissociation are the key points for ORR on layered SiC sheets, the origin of the interaction between O2 and the single-layer SiC is investigated by considering their electronic structure changes. Fig. S9 of supporting information illustrates the spin-polarized partial density of states (PDOS) of O-O bond on the single-layer SiC and Pt(111) surface. Generally, the 5σ, 1π and 2π* orbitals of O2 dominate the adsorption and all of them are broadened due to the interaction with the single-layer SiC and Pt(111) surface. The antibonding 4σ* state is not involved in the adsorption because its position is far below the Fermi level. The antibonding 2π* orbital of gas O2 is partially filled. After adsorption on the single-layer SiC and Pt(111) surface, the initial empty spin-down component of 2π* becomes partially occupied, inducing proportional elongation of the O-O bond length and weakening of O-O bond strength44,45. The electron occupying the antibonding 2π* orbital of adsorbed O2 on the single-layer SiC is 1.55 e compared with 1.19 e on Pt(111) surface and 1.07 e in gas phase according to integrated DOS. Thus, the activation energy of O2 on the single-layer SiC are smaller than that on Pt(111) surface.
In summary, our DFT calculations suggest that novel metal-free layered SiC sheets with N = 1 ~ 3 can exist stably and possess potential ORR catalytic activity due to two advantages of layered SiC sheets compared to Pt(111) surface: (i) free from CO poisoning and (ii) lower activation energies for the RDS of ORR on layered SiC sheets in alkaline media than that on Pt(111) surface. In addition, the corresponding electronic structures are analyzed. The results show that the layered SiC sheets are candidates for practical applications in fuel cells.
Methods
All calculations are performed within density functional theory (DFT) framework as implemented in DMol3 code46,47. The All Electron Relativistic core treat method is implemented for relativistic effects, which explicitly includes all electrons and introduces some relativistic effects into the core. The generalized gradient approximation (GGA) with Perdew−Burke−Ernzerhof (PBE) functional is employed to describe exchange and correlation effects48. PBE is the most common density functional in materials and surface science. However, it cannot accurate describe the van der Waals forces49,50,51,52. This is mainly because the GGA-PBE fails to describe non-local dispersion forces, which are expected to be relevant in layered inorganic compounds and weak adsorption systems. The lack of inclusion of long range correlation in the local density functional (LDF) calculations prevents the accurate calculation of the van der Waals bonds46. Therefore, a Grimme approach is adopted for dispersion corrections. Dmol3 uses a double set of numerically tabulated basis functions46. A more precise term would be “double numerical basis” to be contrasted with double zeta basis, where the radial functions are defined as Slater zeta functions. The basis set can be significantly improved by adding higher angular momentum valence polarization functions and also by core polarization functions46. Total energy for the double basis is quite uniformly higher than the ones for the extended basis sets. It has shown that the basis set produces errors in self-consistent eigenvalues and total energy of ≈0.00003 Ha. In this work, the double numerical atomic orbital augmented by a polarization p-function (DNP) is chosen as the basis set46. The accuracy of DNP is comparable to a Gaussian 6-31(d) basis53. The double basis set may be considered as a large basis especially for the larger molecules. Recent theoretical works based this basis set have shown excellent consistency with experiments54,55. We have also compared our results with that obtained based on triple numerical plus polarization (TNP), as shown in Tables S6 and S7. It can be found that the adsorption energies based on TNP are slightly larger than that based on DNP, while the activation energies and reaction energies of ORR in LH mechanism based on both basis sets are almost the same. A smearing of 0.005 Ha (1 Ha = 27.21 eV) to the orbital occupation is applied to achieve accurate electronic convergence. The spin-unrestricted method is used for all calculations. To ensure high-quality results, the real-space global orbital cutoff radius is chosen as high as 4.6 Å in the computations. The k-point density is set as 2/3 × 2/3 × 1 for per unit cell. The convergence tests for k-point density are shown in Table S8 of supporting information, which shows that the energy of single SiC layer is converging when k-point density is larger than 2/3 × 2/3 × 1. The convergence tolerance of energy is 1.0 × 10−5 Ha, maximum force is 0.002 Ha/Å and maximum displacement is 0.005 Å in the geometry optimization. The transition states for ORR elemental steps are obtained by LST/QST tools in DMol3 code. Frequency calculations are performed to confirm the location of the transition states. A conductor-like screening model (COSMO) is used to simulate a H2O solvent environment throughout the whole process. All data, except for clearly stated, are obtained under this method. The COSMO is a continuum model in which the solute molecule forms a cavity within the dielectric continuum of permittivity56. The DMol3/COSMO method has been generalized to the periodic boundary cases. The deviation of this COSMO approximation from the exact solution is small. For strong dielectrics like H2O, it is less than 1%. The dielectric constant is set as 78.54 for H2O solvent. Our previously theoretical work, focused on ORR on N doped CNT based on COSMO, is consistent with experiments well29. In addition, some other results also show that this implicit solvation model is an effective method to describe the solvation30,31. The solvation energies of ORR intermediates on SiC sheets are shown in Table S9 of supporting information and adsorption energies in a gas phase environment are shown in Table S5 of supporting information, from which we can find that O2 and OH are stabilized on SiC sheets by solvation. For layered SiC sheets, all simulations are performed in a 3 × 3 supercell except for coverage examination of O2 adsorption, where 2 × 2 and 4 × 4 supercells are used. The minimal distance between the SiC layers and their mirror images is set as 15 Å, which is sufficiently large to avoid the interaction between them. The Si-C bond length is 1.79 Å. The Pt(111) surface is modeled by a periodic array of Pt slabs with a vacuum width in excess of 15 Å. The p(3 × 3) unit cells with three layer Pt slabs are used throughout in the ORR calculations. The bottom two Pt layers are fixed at their bulk positions and the top Pt layer is allowed to relax fully. Tests performed with four and five atomic layers, top two of which are allowed to relax, do not show significant differences in structural parameters. As shown in Table S10 of supporting information, the adsorption energy variation for adsorbed molecules is smaller than 0.09 eV.
In this work, both LH and ER mechanisms are expected to proceed. The LH reaction referrs to the reaction of adsorbed hydrogen atoms with another adsorbate and the ER reaction does the reaction of a proton from solution interacting with an adsorbate. The complete electrochemical ORR in ER mechanism involves four CPETs to O2 molecule at the cathode31,39. Electron transfers are coupled with a proton transfer as well. Barriers for electrochemical proton-transfer have been calculated for the reduction of O2 to OOH and OH to H2O on Pt57,58. In both cases, the proton-transfer reaction barrier calculated is small (0.15 eV to 0.25 eV) at the low potential that elementary steps are exothermic and diminishes with higher applied voltages57,58. Similarly, as a first approximation, we expect also that barriers for electrochemical proton transfers to adsorbed species will be small and be easily surmountable at room temperature. As a result, we only calculated reaction energy for ORR in the ER mechanism. Free energies of the ORR intermediates are calculated based on a computational hydrogen electrode (CHE) model suggested by Nøskov et al40,59. Free energy change (ΔG) is determined by ΔG = ΔE + ΔZPE − TΔS + ΔGpH + ΔGU, where ΔE is the reaction energy, ΔZPE is the zero point energy, T is temperature and ΔS is the change in the entropy. ΔGpH and ΔGU are the free energy contributions due to variations in H+ concentration and electrode potential, U, respectively. In this work, we consider the contributions of ΔE, ΔGpH and ΔGU to free energy and neglect the effects of other terms60. Effects of other terms will be included in a forthcoming publication. We assume pH = 0, 1, 3 and 5 for acidic medium and pH = 9, 11 and 14 for alkaline medium. The pH effect is very hard to be considered directly in the electrolyte. Generally, it can be treated following the method directed by Nørskov et al40. At a pH differing from 0, the free energy of H+ ions can be corrected by the concentration: GpH = −kBTln[H+] = pH × kBTln10. This pH-dependence effect does not enter the COSMO-approach.
Naturally, ORR is a highly complicated process. Undertaking a kinetics analysis using rate constants derived from first-principles calculations would be an ideal way to determine which pathways are relevant under different conditions. Such a kinetics analysis is unfortunately difficult, since it would require rigorous double-layer effects, such as intermediate concentrations, surface coverages, or the potential drop within the interface. At here, the potential dependent rate constants k(U) are obtained based on Eyring's canonical transition state theory:  , where kB is Boltzmann constant, T = 298.15 K, h is Planck constant and ΔG(U) is the potential-dependent barrier for that process39,42,43.
, where kB is Boltzmann constant, T = 298.15 K, h is Planck constant and ΔG(U) is the potential-dependent barrier for that process39,42,43.
References
Gasteiger, H. A., Kocha, S. S., Sompalli, B. & Wagner, F. T. Activity benchmarks and requirements for Pt, Pt-alloy and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B 56, 9–35 (2005).
Yu, X. & Ye, S. Recent advances in activity and durability enhancement of Pt/C catalytic cathode in PEMFC part II: degradation mechanism and durability enhancement of carbon supported platinum catalyst. J. Power Sources 172, 145–154 (2007).
Stamenkovic, V. R. et al. Trends in electrocatalysis on extended and nanoscale Pt-Bimetallic alloy surfaces. Nat. Mater. 6, 241–247 (2007).
Wang, C. et al. Rational development of ternary alloy electrocatalysts. J. Phys. Chem. Lett. 3, 1668–1673 (2012).
Wang, C., Markovic, N. M. & Stamenkovic, V. R. Advanced platinum alloy electrocatalysts for the oxygen reduction reaction. ACS Catal. 2, 891–898 (2012).
Greeley, J. et al. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 1, 552–556 (2009).
Strasser, P. et al. Lattice-strain control of the activity in dealloyed core-shell fuel cell catalysts. Nat. Chem. 2, 454–460 (2010).
Karan, H. I. et al. Catalytic activity of platinum monolayer on Iridium and Rhenium alloy nanoparticles for the oxygen reduction reaction. ACS Catal. 2, 817–824 (2012).
Srivastava, R., Mani, P., Hahn, N. & Strasser, P. Efficient oxygen reduction fuel cell electrocatalysis on voltammetrically dealloyed Pt–Cu–Co nanoparticles. Angew. Chem. Int. Ed. 46, 8988–8991 (2007).
Kongkanand, A., Kuwabata, S., Girishkumar, G. & Kamat, P. Single-wall carbon nanotubes supported platinum nanoparticles with improved electrocatalytic activity for oxygen reduction reaction. Langmuir 22, 2392–2396 (2006).
Guo, S. & Sun, S. FePt nanoparticles assembled on graphene as enhanced catalyst for oxygen reduction reaction. J. Am. Chem. Soc. 134, 2492–2495 (2012).
Zhang, Y. et al. Nanoporous graphitic-C3N4@carbon metal-free electrocatalysts for highly efficient oxygen reduction. J. Am. Chem. Soc. 133, 20116–20119 (2011).
Yang, S., Feng, X., Wang, X. & Müllen, K. Graphene-based carbon nitride nanosheets as efficient metal-free electrocatalysts for oxygen reduction reactions. Angew. Chem. Int. Ed. 50, 5339–5343 (2011).
Yu, D., Zhang, Q. & Dai, L. Highly efficient metal-free growth of nitrogen-doped single-walled carbon nanotubes on plasma-etched substrates for oxygen reduction. J. Am. Chem. Soc. 132, 15127–15129 (2010).
Liu, Z. W. et al. Phosphorus-doped graphite layers with high electrocatalytic activity for the O2 reduction in an alkaline medium. Angew. Chem. Int. Ed. 50, 3257–3261 (2011).
Choi, C. H., Park, S. H. & Woo, S. I. Binary and ternary doping of nitrogen, boron and phosphorus into carbon for enhancing electrochemical oxygen reduction activity. ACS Nano 6, 7084–7091 (2012).
Cruz-Silva, E. et al. Electronic transport and mechanical properties of phosphorus- and phosphorus-nitrogen-doped carbon nanotubes. ACS Nano 3, 1913–1921 (2009).
Zhang, L. & Xia, Z. Mechanisms of oxygen reduction reaction on nitrogen-doped graphene for fuel cells. J. Phys. Chem. C 115, 11170–11176 (2011).
Gong, K., Du, F., Xia, Z., Durstock, M. & Dai, L. Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 323, 760–764 (2009).
Yang, L. et al. Boron-doped carbon nanotubes as metal-free electrocatalysts for the oxygen reduction reaction. Angew. Chem. Int. Ed. 50, 7132–7135 (2011).
Yang, Z. et al. Sulfur-doped graphene as an efficient metal-free cathode catalyst for oxygen reduction. ACS Nano 6, 205–211 (2011).
Sun, X. J. et al. A class of high performance metal-free oxygen reduction electrocatalysts based on cheap carbon blacks. Sci. Rep. 3, 2505 (2013).
Zhang, Y. et al. Manageable N-doped graphene for high performance oxygen reduction reaction. Sci. Rep. 3, 2771 (2013).
Hu, X., Wu, Y., Li, H. & Zhang, Z. Adsorption and activation of O2 on nitrogen-doped carbon nanotubes. J. Phys. Chem. C 114, 9603–9607 (2010).
Ikeda, T. et al. Carbon alloy catalysts: active sites for oxygen reduction reaction. J. Phys. Chem. C 112, 14706–14709 (2008).
Lin, S. S. Light-emitting two-dimensional ultrathin silicon carbide. J. Phys. Chem. C 116, 3951–3955 (2012).
Eichler, A. & Hafner, J. Molecular precursors in the dissociative adsorption of O2 on Pt(111). Phys. Rev. Lett. 79, 4481–4484 (1997).
Eichler, A., Mittendorfer, F. & Hafner, J. Precursor-mediated adsorption of oxygen on the (111) surfaces of platinum-group metals. Phys. Rev. B 62, 4744–4755 (2000).
Zhang, P., Lian, J. S. & Jiang, Q. Potential dependent and structural selectivity of the oxygen reduction reaction on nitrogen-doped carbon nanotubes: a density functional theory study. Phys. Chem. Chem. Phys. 14, 11715–11723 (2012).
Sha, Y., Yu, T. H., Liu, Y., Merinov, B. V. & Goddard, W. A. Theoretical study of solvent effects on the platinum-catalyzed oxygen reduction reaction. J. Phys. Chem. Lett. 1, 856–861 (2010).
Keith, J. A., Jerkiewicz, G. & Jacob, T. Theoretical investigations of the oxygen reduction reaction on Pt(111). ChemPhysChem 11, 2779–2794 (2010).
Thorum, M. S., Hankett, J. M. & Gewirth, A. A. Poisoning the oxygen reduction reaction on carbon-supported Fe and Cu electrocatalysts: evidence for metal-centered activity. J. Phys. Chem. Lett. 2, 295–298 (2011).
Zhang, P., Chen, X. F., Lian, J. S. & Jiang, Q. Structural selectivity of CO oxidation on Fe/N/C catalysts. J. Phys. Chem. C 116, 17572–17579 (2012).
Blyholder, G. Molecular orbital view of chemisorbed carbon monoxide. J. Phys. Chem. 68, 2772–2777 (1964).
Mazzone, G., Rivalta, I., Russo, N. & Sicilia, E. Interaction of CO with PdAu(111) and PdAu(100) bimetallic surfaces: a theoretical cluster model study. J. Phys. Chem. C 112, 6073–6081 (2008).
Liu, Z. P. & Hu, P. General trends in CO dissociation on transition metal sufaces. J. Chem. Phys. 114, 8244–8247 (2001).
Michaelides, A. et al. Identification of general linear relationships between activation energies and enthalpy changes for dissociation reactions at sufaces. J. Am. Chem. Soc. 125, 3704–3705 (2003).
Bligaard, T. et al. The Brønsted–Evans–Polanyi relation and the volcano curve in heterogeneous catalysis. J. Catal. 224, 206–217 (2004).
Keith, J. A. & Jacob, T. Theoretical studies of potential-dependent and competing mechanisms of the electrocatalytic oxygen reduction reaction on Pt(111). Angew. Chem. Int. Ed. 49, 9521–9525 (2010).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cll cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Lim, D. H. & Wilcox, J. Mechanisms of the oxygen reduction reaction on defective graphene-supported Pt nanoparticles from first-principles. J. Phys. Chem. C 116, 3653–3660 (2012).
Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 3, 107–115 (1935).
Gao, W., Keith, J. A., Anton, J. & Jacob, T. Theoretical elucidation of the competitive electro-oxidation mechanisms of formic acid on Pt(111). J. Am. Chem. Soc. 132, 18377–18385 (2010).
Wang, C. M., Fan, K. N. & Liu, Z. P. Origin of oxide sensitivity in gold-based catalysts: a first principle study of CO oxidation over Au supported on monoclinic and tetragonal ZrO2 . J. Am. Chem. Soc. 129, 2642–2647 (2007).
Ou, L., Yang, F., Liu, Y. & Chen, S. First-principle study of the adsorption and dissociation of O2 on Pt(111) in acidic media. J. Phys. Chem. C 113, 20657–20665 (2009).
Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 92, 508–517 (1990).
Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 113, 7756–7764 (2000).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Graziano, G., Klimeš, J., Fernandez-Alonso, F. & Michaelides, A. Improved description of soft layered materials with van der Waals density functional theory. J. Phys.: Condens. Matter 24, 424216 (2012).
Carrasco, J., Klimeš, J. & Michaelides, A. The role of van der Waals forces in water adsorption on metals. J. Chem. Phys. 138, 024708 (2013).
Björkman, T., Gulans, A., Krasheninnikov, A. V. & Nieminen, R. M. van der Waals bonding in layered compounds from advanced density-functionals first-principles calculations. Phys. Rev. Lett. 108, 235502 (2012).
Schimaka, L. et al. Accurate surface and adsorption energies from many-body perturbation theory. Nat. Mater. 9, 741–744 (2010).
Liu, P. & Rodriguez, J. A. Catalysts for hydrogen evolution from the [NiFe] hydrogenase to the Ni2P(001) surface: the importance of ensemble effect. J. Am. Chem. Soc. 127, 14871–14878 (2005).
Rodriguez, J. A. et al. Activity of CeOx and TiOx nanoparticles grown on Au(111) in the water-gas shift reaction. Science 318, 1757–1760 (2007).
Wu, Z. et al. Structure sensitivity of low-temperature NO decomposition on Au surfaces. J. Catal. 304, 112–122 (2013).
Klamt, A. & Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. 2, 799–805 (1993).
Janik, M. J., Taylor, C. D. & Neurock, M. First-principles analysis of the initial electroreduction steps of oxygen over Pt(111). J. Electrochem. Soc. 156, B126–B135 (2009).
Tripkovic, V., Skúlason, E., Siahrostami, S., Nørskov, J. K. & Rossmeisl, J. The oxygen reduction reaction mechanism on Pt(111) from density functional theory calculations. Electrochim. Acta 55, 7975–7981 (2010).
Yu, L., Pan, X., Cao, X., Hu, P. & Bao, X. Oxygen reduction reaction mechanism on nitrogen-doped graphene: a density functional theory study. J. Catal. 282, 183–190 (2011).
Roudgar, A., Eikerling, M. & van Santen, R. Ab initio study of oxygen reduction mechanism at Pt4 cluster. Phys. Chem. Chem. Phys. 12, 614–620 (2010).
Acknowledgements
This work was financially supported by National Key Basic Research and Development Program (Grant No. 2010CB631001), Natural Science Foundation of Jiangsu (No. SBK201341900), China Postdoctoral Science Foundation (No. 2013M541611) and the Senior Intellectuals Fund of Jiangsu University (No. 12JDG094 and 13JDG032). We acknowledge the supports from High Performance Computing Center (HPCC) of Jilin University.
Author information
Authors and Affiliations
Contributions
P.Z. conceived the initial idea of this research, performed the computer simulations and wrote the paper. B.B.X., X.L.H., Y.F.Z. and Q.J. participated in the discussion and revised the manuscript. Y.F.Z. and Q.J. guided the work.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Layered SiC Sheets: A Potential Catalyst for Oxygen Reduction Reaction
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Zhang, P., Xiao, B., Hou, X. et al. Layered SiC Sheets: A Potential Catalyst for Oxygen Reduction Reaction. Sci Rep 4, 3821 (2014). https://doi.org/10.1038/srep03821
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03821
This article is cited by
-
OH-Functionalized N-Doped Graphene Quantum Dots as an Efficient Metal-Free Catalysts for Oxygen Reduction Reaction in PEMFCs
Electrocatalysis (2024)
-
Heteroatom doping regulates the catalytic performance of single-atom catalyst supported on graphene for ORR
Nano Research (2024)
-
Catalytic activity of OH functionalized N-doped graphene in oxygen reduction reaction for fuel cell applications: a DFT study
Applied Physics A (2023)
-
Unveiling the effect of 2D silagraphene structural diversity on electronic properties: DFT, DOS, and ELF studies
Journal of Molecular Modeling (2022)
-
Advanced Noncarbon Materials as Catalyst Supports and Non-noble Electrocatalysts for Fuel Cells and Metal–Air Batteries
Electrochemical Energy Reviews (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
