
Abstract
A structure-switching DNA optical biosensor for rapid on-site/in situ detection of heavy metal ions is reported. Mercury ions (Hg2+), highly toxic and ubiquitous pollutants, were selected as model target. In this system, fluorescence-labeled DNA containing T-T mismatch structure was introduced to bind with DNA probes immobilized onto the sensor surface. In the presence of Hg2+, some of the fluorescence-labeled DNAs bind with Hg2+ to form T-Hg2+-T complexes through the folding of themselves into a hairpin structure and dehybridization from the sensor surface, which leads to decrease in fluorescence signal. The total analysis time for a single sample was less than 10 min with detection limit of 1.2 nM. The rapid on-site/in situ determination of Hg2+ was readily performed in natural water. This sensing strategy can be extended in principle to other metal ions by substituting the T-Hg2+-T complexes with other specificity structures that selectively bind to other analytes.
Similar content being viewed by others
Introduction
Heavy metal pollution in natural water environments worldwide is an urgent problem because of the severe threat it poses to human health and the environment. This concern has led to strict regulations on the maximum metal concentrations allowed in natural waters1. Traditional quantitative methods, such as atomic absorption/emission spectroscopy2, inductively coupled plasma mass spectrometry3 and cold vapor atomic fluorescence spectrometry (CVAFS)4,5, have been extensively applied to detect heavy metal ions with high sensitivity. In addition to the expensive and sophisticated instrumentation required, these techniques normally involve complicated chemical processes for extracting metal ions from the as-sampled water, in which the speciation change of metal ions is unavoidable5,6,7. Several studies have reported that the uptake of heavy metals by organisms does not depend entirely on the total metal concentration in solution. Better correlations have been found between metal uptake and free metal ion concentration or labile metal concentration8,9. Save for some exceptions, free metal ion concentration, in most cases, ultimately determines the bioavailability and toxicity of heavy metals10,11. However, large-scale determination of heavy metals can be costly, time consuming and labor intensive. To protect the environment and human health, as well as to provide rapid and inexpensive characterization and remediation of these sites, a portable, low-cost and fast heavy metal analysis system for initial on-site/in-situ screening of heavy metal-contaminated sites should be prioritized.
Recently, much effort has been focused on the design of DNA-based sensors to detect metal ions based on the ability of some metal ions that selectively bind to some bases to form stable metal-mediated DNA duplexes12,13. For example, mercury ions (Hg2+) are capable of selectively coordinating thymine (T) bases to form stable T-Hg2+-T complexes14 and Ag+ interacts specifically with cytosine-cytosine (C-C) mismatches15. For Pb2+ detection, most sensors are based on the Pb2+-dependent DNAzyme and the Pb2+-stabilized G-quadruplex16,17. Accordingly, various detection techniques adopting fluorescence, surface-enhanced Raman spectroscopy, resonance scattering, colorimetry and electrochemical methods were applied to selectively detect these heavy metal ions based on structure-switching DNA12,13,14,15,16,17. Structure-switching DNA biosensors have several unique characteristics18. First, structure switching is induced by the formation of many weak non-covalent bonds, which is generally specific to a given ligand-biomolecule interface and mostly insensitive to other molecules even in complex environments (e.g., natural water, living cells, or blood serum). Second, given that the switching (signal transduction) is rapid, reversible and reagent-free, these nanoscale switches are suited for rapid, real-time and in situ monitoring of specific targets. Finally, the conformational equilibria of structure-switching DNA are related to both its concentration and thermodynamics, which renders structure-switching DNA biosensors quantitative and their dynamic ranges rationally optimized without altering their binding specificity. To date, numerous biosensors for the detection of heavy metal ion detection based on DNA structure switching have been reported. However, most of these detections were performed based on the homogeneous reaction principle and sophisticated instrumentations are required, which made them unsuitable for rapid on-site/in-situ detection of heavy metal ions12,13,14,15,16,17,18.
Evanescent wave fiber optic biosensors have been conveniently and effectively used to determine various trace amounts of targets based on the principle of total internal reflection fluorescence (TIRF)19. These biosensors possess several promising advantages, such as small size, good flexibility, multiplexing capability, low propagation loss and resistance to electromagnetic interference. When light propagates through a fiber optic on the basis of TIR, a very thin electromagnetic field (known as the “evanescent wave”) is generated. This field decays exponentially with distance from the interface, with a typical penetration depth of a few nanometers to several hundred nanometers. This evanescent wave can excite fluorescence that approach the sensing surface, such as those used for fluorescence labeling of molecules (e.g., DNA or antibody) as they bind to the optical sensor surface. The limited range of the evanescent wave allows the distinction between bound and unbound fluorescent complexes. Thus, real-time detection of the surface reaction of fiber optic followed by on-site/in situ measurement collection is possible.
Combined with the advantages of an evanescent wave optical biosensor and structure-switching DNA, an evanescent wave all-fiber biosensor system that is suitable for rapid on-site/in-situ detection of heavy metal ions is proposed in this study. Hg2+, a highly toxic and ubiquitous pollutant that affects human and ecosystem health, was selected as model target. Given the bioaccumulative property of Hg2+, long-term exposures to even minute amounts of this metal can result in a number of severe health problems, such as brain damage, kidney failure, chromosome breakage and severe neurological disorders. The World Health Organization suggests 30 nM of inorganic mercury as the guideline tolerable value for mercury in drinking water, whereas the United States Environmental Protection Agency (USEPA) guideline value is 10 nM20. In our newly developed heavy metal-sensing system, a short DNA probe that is complementary to the fluorescence-labeled complementary DNA (cDNA) sequence containing a T-T mismatch structure was first immobilized onto fiber optical sensing surface. To selectively detect Hg2+, fluorescence-labeled cDNA was introduced to bind with the immobilized DNA probes onto the sensor surface. The prepared fiber optic biosensor was introduced into the Hg2+ solution. Some of the fluorescence-labeled DNAs bind with Hg2+ to form T-Hg2+-T complexes through the folding of themselves into a hairpin structure and dehybridization from the fiber optic probe, which leads to a decrease in fluorescence signal. Higher Hg2+ concentration causes more fluorescence-labeled cDNAs to dehybridize from the sensor surface, thus resulting in the detection of lower fluorescence signals. The binding kinetics of Hg2+ and fluorescence-labeled DNAs containing a T-T mismatch structure, as well as the sensitivity and selectivity of this biosensor based on structure-switching DNA, were also evaluated. To our best knowledge, this is the first optical biosensor for rapid on-site/in-situ detection of heavy metal ions based on structure-switching DNA.
Results
Sensing mechanism for heavy metal ion detection based on structure-switching DNA
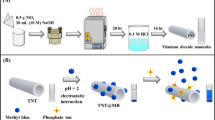
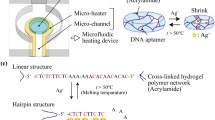
The heavy metal sensing mechanism in the evanescent wave optical biosensing platform (Figure 1) through the “turn off” model based on structure-switching DNA is depicted in Figure 2. The fluorescence-labeled T-rich cDNA initially contained a short sequence structure of 10 bps (shown in green and red in Figure 2) at the 5′ end of the cDNA. This structure is complementary and therefore, hybridize with the DNA probe (shown in blue in Figure 2) that is immobilized onto the fiber optic sensor surface. Then, Hg2+ was then introduced into the sensor surface. The cDNA probe also contained a structure of T-T mismatch pairs, which can bind with Hg2+ to form the T-Hg2+-T complex by the folding of the cDNA probe segments (shown in green in Figure 2) into a hairpin-like geometry. This structure triggers the release of the fluorescence-labeled cDNA, resulting in a decreasd fluorescence signal. The higher the concentration of Hg2+, the more cDNA is released, thereby resulting in lower fluorescence signal. Finally, the excess fluorescence-labeled cDNA can be removed completely by adding regeneration solution (0.5% SDS, pH 1.9) and the fiber optic sensor can be reused for subsequent test.

(A) Schematic of evanescent wave all-fiber optical biosensing platform. (B) Photograph of the biosensing platform for the on-site/in situ detection of heavy metal ions. (Photo by Feng Long).

Schematic of structure-competitive sensing mechanism of Hg2+ detection.
Figures 3A and 3B show the exemplary fluorescence signal profile developed during a typical test cycle for Hg2+ detection using the optical sensor scheme, which include the following processes: introduction of the fluorescence-labeled cDNA and binding of cDNA and DNA probe immobilized onto the sensor surface (Figure 3, phase I); introduction of Hg2+ and reaction between cDNA and Hg2+ (Figure 3, phase II); and sensor regeneration (Figure 3, phase III). First, 0.3 mL of 20 nM fluorescence-labeled cDNA was introduced onto the fiber optic sensor surface. As shown in phase I in Figure 3B, the fluorescence signal exponentially increased over time and eventually reached a plateau after 2 min. Real-time monitoring of the fluorescence signal reflected the kinetic rates of the binding reaction that occurred between the Cy5.5-cDNA and the immobilized DNA probe. Meanwhile, the photobleaching rate of the bound fluorophores is negligible even when the laser is turned on throughout the entire test cycle (Figure 3, phase I).

Exemplary signal profiles for Hg2+ detection with the structure-switching DNA-based optical biosensor.
(A) Experiment with 30 nM fluorescence-labeled non-specific DNA sequence as control. (B) Experiment with 20 nM fluorescence-labeled cDNA that specifically binds to DNA probe immobilized onto the sensor surface and with 1 × PBS introduced. (C) Experiment with the mixture of 20 nM fluorescence-labeled cDNA that specifically binds to DNA probe immobilized onto the sensor surface and 20 nM Hg2+ introduced.
We conducted a control experiment to evaluate the results of the DNA probe immobilization and to confirm that the observed fluorescence signal was produced by the specific DNA hybridization between cDNA with the complementary sequence of the DNA probe on the sensor. The results of the control experiment are shown in Figure 3. No detectable fluorescence intensity was observed when the fluorescence-labeled non-complementary DNA (30 nM) [5′-Cy5.5-ATGCTCCCGAGA-3′] (Takara Biotechnology (Dalian) Co., China) was delivered over the sensing surface, indicating that no non-specific DNA adsorption or hybridization occurred on the sensor surface. Furthermore, the free Cy5.5-cDNA in the bulk solution was less excited by the evanescent wave and the contribution of its fluorescence signal was negligible. This result contributed to the decay length of the evanescent field on the sensor surface in the nanometer scale. Light intensity was only concentrated on the molecules that covered the sensor surface and the unwanted signal from the bulk water can be excluded. When 20 nM cDNA was introduced, a signal to noise (S/N) ratio (the ratio of the maximum fluorescent signal to the base line) of more than 5 was obtained (Figure 3). These results demonstrated the high sensitivity of the sensor for detecting specific DNA hybridization and the rapid completion of the hybridization process.
In the absence or presence of Hg2+, 0.3 mL of samples was delivered onto the sensor surface (Figures 3B and 3C, phase II). In the absence of Hg2+, only a little cDNA dehybridized from the sensor surface and the fluorescence signal slightly decreased. When Hg2+ concentration increased, more cDNA dehybridized from the sensor surface, therefore resulting in lower fluorescence signals. The fluorescence signal exponentially decreased over time and eventually reached a plateau after 2 min. Thus, considering the required signal intensity level and the aim to shorten the assay time, a reaction time of 2 min was chosen. For each assay, the net fluorescence intensity (I) used for the subsequent determination of dose-response relationships was calculated according to Equation 1:


The response was normalized according to the following equation:

where Ib is the net fluorescent intensity at the peak value by subtracting the baseline value.
Finally, the sensing surface was regenerated with 0.5% SDS solution (pH 1.9) for 90 s and was washed with PBS solution (Phase III in Figure 3B and 3C) to reuse the sensor. Complete cDNA removal from the sensor surface was achieved. To test the next sample, cDNA was introduced again onto the sensor surface. The fluorescence signal trace was nearly the same as the preceding one, indicating that the immobilized recognition element has good regeneration performance. The immobilized recognition element could actually sustain at least 50 successive assays without any significant decrease in performance (less than 5% decrease).
Optimization of fluorescence-labeled DNA concentration
After the sensing mechanism was established, DNA probe concentration was optimized to improve assay sensitivity. To determine the optimum experimental condition, a sensitivity index (F/F0), the ratio between the net fluorescence intensity obtained in the presence of the analyte (F = Ib − Is) and that in the absence of the analyte (F0 = Ib − I0), was introduced. In this study, the sensitivity index was calculated from the fluorescence intensity with 5 nM Hg2+ (F) and in the absence of target metal ions (F0). For the practical application of the sensor in the desired dynamic range, the most acceptable cDNA probe concentration was chosen according to the following criteria: (1) the S/N ratio should be as high as possible and preferably larger than 5 to generate a reasonable fluorescence intensity and (2) the sensitivity index should be as low as possible and favorably below 0.90. Based on these criteria, several experiments were conducted (Figure 4) and the optimum concentration of fluorescence-labeled cDNA probe was found to be 20 nM. Lower concentrations of fluorescence-labeled cDNA probe results in effective analyte competition in structure-switching bioassay, which leads to better sensitivity.

S/N ratio (magenta) and sensitivity index (blue) change in relation to the concentration of cDNA.
Dose-response curves of Hg2+
After the general assay parameters were optimized, Hg2+ standard solutions in the range of 0 nM to 1000 nM were analyzed to verify the DNA structure-switching based bioassay. Figure 5A shows the temporal fluorescence signal during a typical test cycle for Hg2+ detection using the all-fiber optical biosensing platform, including the introduction and hybridization of the fluorescence-labeled cDNA, the introduction and reaction of sample containing different Hg2+ concentrations and the washing step. Figure 5A also shows the decrease in the kinetics of the fluorescence with the introduction of Hg2+ to the sensing interface and the proportional decrease in the fluorescence signal was induced by the increase in Hg2+ concentration.

(A) Exemplary sensor response curves and the signals obtained with various concentration of Hg2+. (B) Logarithmic calibration plot for determination of Hg2+ using structure-switching DNA based optical biosensor system.
Figure 5B shows the calibration curves (the average of three independent curves) for Hg2+, which were normalized by expressing the signal of each standard point as the ratio of that of the blank sample containing no Hg2+. The signal intensities were fitted to the four-parameter logistic equation21. The error bars in the figure correspond to the standard deviation of the data points in triplicate experiments, with the standard deviation of all the data points within 7.8%. With the dose-response curve and the S/N ratio of 5.1, the detection limit was determined as approximately 1.2 nM based on the average standard deviation of the measurements (σ) and the slope of the dose-response fitting curve (S) as 3σ/S22. Therefore, this novel sensor can be applied to the direct detection of Hg2+ in natural water with the ability to meet even the most stringent requirements demanded by the USEPA (10 nM). In our experiment, the detection limit is initially comparable with that of other sensing assay methods, such as the “turn-on” fluorescence sensor (3.2 nM)23, ratiometric sensor (50 nM)24, electrochemical sensor based on gold nanoparticle (Au NP) amplification (0.5 nM)25 and Au NP-enhanced SPR sensor (5 nM)26. Recently, a plasmon-enhanced infrared spectroscopy was shown to exhibit a very low detection limit of 37 ppt27. To the best of our knowledge, this is the first report of such a high sensitivity for the detection of Hg2+ using an evanescent wave optical biosensor. The high sensitivity of this biosensor is possibly due to the surface chemistry of the aminated DNA on the probe surface that maintained the activity of the immobilized DNA probe toward Hg2+ and cDNA and diminished non-specific adsorption. Compared with abovementioned sensors, this portable optical biosensor based on structure-switching DNA detection mode is much simpler and time-saving (operation time was only 10 min, including measurement and regeneration). Furthermore, in the present system, the sensor probe can be regenerated without comprising bioreactivity for multiple analyses and can easily be extended for the determination of other heavy metal ions or small molecular analytes for which specific sensing probes are available.
Selectivity of the sensing system
To assess the selectivity of the sensor, its responses to potential interference from other metal cations, such as Mn2+, Mg2+, Ca2+, Co2+, Zn2+, Fe2+, Cu2+, Sn2+, Cr2+, Ni2+ and Pb2+, at concentrations up to 20 μM were evaluated. As indicated in Figure 6, the sensor exhibits high selectivity toward Hg2+ with no significant response to the other divalent metal ions. Moreover, Hg2+ and other metal ions were combined to form a metal ion soup as a sample for the anti-jamming capability testing of the sensor. The fluorescence signal was obviously lower than that of other samples without Hg2+. These results clearly indicated that the approach is not only insensitive to other metal ions but also selective toward Hg2+ in their presence. As noted above, the present sensor had excellent anti-jamming capability and outstanding selectivity. This selectivity must be due to its selective binding of T-T mismatches, resulting in the formation of stable T-Hg2+-T complexes.

Selectivity of the structure-switching DNA-based Hg2+ biosensor.
On-site and in-situ measurement of Hg2+
To further evaluate the on-site and in-situ detection ability of our developed system, one microcosm was constructed as described in the methods section and the results are shown in Figure 7. Results show that although the initial Hg2+ concentration was about 500 nM, the adsorption process of sludge for Hg2+ was very quick28 and the concentration of the first sample was only 350 and 320 nM by CVAFS and biosensor, respectively. Then, the Hg2+ concentration rapidly decreased with time. The adsorption reaction between Hg(II) and the sludge surface sites can be expressed as

We assume that the resulting time dependent surface adsorption Θ follows the Langmuir equation and can be described by

where kon is the adsorption rate constant, koff is the desorption rate constant and C is the concentration of Hg(II) adsorbed by the sludge at time t. At the onset of adsorption, the adsorption rate is at the maximum and the desorption rate and Θ are zero. In Figure 7A, the fitting began with an estimation of the parameter values, which were continually optimized until the residual sum of squares no longer decreased significantly. The experimental data and the modeled curve (Figure 7A, red line) derived from pseudo-second order equation was in good agreement.

(A) On-site/in situ detection of Hg2+ in the microcosm by optical biosensor and validation by CVAFS. (B) Linear correlation between the optical biosensor and CVAFS.
A comparison between the methods yielded good correlation results (y = 0.9623X − 14.0111), with the slope (0.9623) of the linear regression equation indicating that with respect to the CVAFS measured values, the biosensor results were somewhat underestimated. Nevertheless, the correlation coefficient (r2 = 0.9966) demonstrated satisfactory agreement between the methods and the in-situ detection value had no obvious difference with the on-site detection result. These results imply that this proposed biosensor method could be used for the portable on-site/in-situ determination of heavy metal ions in the field of environmental monitoring. This is especially important when optical biosensor techniques are intended to provide bioavailability information such that the measurement of the heavy metal contamination levels can be obtained without any sample modification caused by changes in pressure, temperature, or sample contamination.
Discussion
This is first experiment that presented a structure-switching DNA based biosensor for rapid on-site/in situ detection of heavy metal ions. The primary advantages of this structure-switching DNA-based biosensor include sensitivity, speed, portability and minimum sample manipulation. We have shown that field-based biosensor analyses can be accomplished with good correlation to laboratory-based conventional analytical methods. This initial proof-of-concept study has clearly shown that on-site/in situ measurements obtained with such a structure-switching DNA-based sensor can be used to provide rapid on-site/in situ information on these heavy metals in natural water. Although we have only demonstrated the Hg2+ detection, this sensing strategy can be extended, in principle, to any substance, such as other metal ions or small molecules, by substituting the T-Hg2+-T complexes with other specificity structures that selectively bind other analytes. Coupled with the remote-detection ability of fiber optic sensors, this sensing strategy may be an alternative method for the analysis and assessment of the transport and fate of heavy metal ions or other small molecules in environmental samples.
Methods
Instrumentation: evanescent wave all-fiber biosensing platform
An all-fiber biosensing platform for the detection of the fluorescence signal is shown in Figure 1. In this system, the 635 nm 15 mW pulse diode laser (HuayuanStar Ltd., China) with pigtail was selected as the excitation light resource because of its monochromaticity, stability and compactness. Instead of using various optical separation components (e.g., lens and dichroic mirror), a single-multi mode fiber optic coupler was utilized to transmit the excitation light and to collect and transmit fluorescence. The laser beam from the diode laser was directly launched into the single-mode fiber of the single multi-mode silica fiber optic coupler. Laser light then entered the multi-mode fiber with a diameter of 600 μm and numerical aperture of 0.22 from the single-mode fiber. Meanwhile, this multi-mode fiber was used to collect the fluorescence excited by the laser. The excitation light from the laser, through the fiber connector, was then coupled to a combination tapered fiber optic sensor, the fabrication and preparation of which was described previously29. The incident light was propagated along the entire length of the fiber optic sensor via TIR. The evanescent wave was generated at the surface of the probe and then interacted with the surface-bound fluorescent-labeled analyte complexes, which excited the fluorophores. A bandpass filter was placed before the photodiodes to ensure that only the fluorescence signal without any residual pump signal was measured. The collected fluorescence was subsequently monitored by photodiodes through lock-in detection. By reducing optical components and unnecessary optical alignment, the biosensor platform can be suitable for on-site and in-situ detection at field sites. Furthermore, light transmission efficiency increased, light loss decreased and S/N ratio improved.
During on-site detection, the fiber optic sensor was embedded in a flow glass cell with a flow channel having a nominal dimension of 60 mm in length and 2 mm in diameter (Figure 1B). All reagents were delivered by a flow delivery system operated using a peristaltic pump. The fluid delivery system control and data acquisition and processing were automatically performed by the embedded computer. During in-situ detection, the fiber optic sensor prepared was directly immersed into the sample.
Preparation of structure-switching DNA biosensor
Details on the modification of the combination tapered fiber optic sensor were described previously29. Briefly, DNA probes were immobilized onto the fiber optic sensor surface procedures that follow. First, the optical fiber sensor was cleaned with the piranha solution (H2SO4/H2O2, 3:1 [v/v]). Second, the sensor was aminated by immersion in 2% (v/v) ATPS acetone solution for 60 min, followed by acetone wash (three times) and ultrapure water wash and dried in an oven for 30 min at 110°C. To immobilize the DNA probe [5′-NH2-(CH2)6-GTACAAACAA-3′] (Takara Biotechnology (Dalian) Co., China) onto the aminated sensor surface, the glutaraldehyde-covalent-coupling strategy was applied. The sensor is immersed in a 5.0% (v/v) glutaraldehyde solution for 1 h at 37°C, washed with water and then immersed overnight in 1 mL of a 0.5 μM DNA probe in PBS solution (pH 7.4) at 4°C. The resultant sensor surface was quenched by dipping in 2 mg/mL ethanolamine for 1 h to block the remaining aldehyde sites. The fluorescence labeled cDNA sequence containing T-T mismatch structure [5′-Cy5.5-CTTTCTTCCCCGTTGTTTGTAC-3′] was introduced into the fiber optic sensor for hybridization, which was used for the Hg2+ detection.
On-site and in situ detection of real water samples
To evaluate the on-site and in situ detecting ability of our biosensor, one microcosm was prepared in 250 mL serum bottles using 2.4 g of sludge (from Lake Beihai's sediments of Beijing, China) and 200 mL pure water. The sludge and water were mixed and deposited for 2 h. Afterward, 2 mL 50 mM Hg2+ was added to the microcosms. In the first two hours, 1 mL supernatant solution was taken for the biosensor on-site detection and CVAFS detection according to USEPA Method 245.7 was performed every 30 mins30. At the third hour, the developed fiber optic sensor was directly immersed into the supernatant solution to detect the Hg2+ concentration in real water samples in situ. Simultaneously, CVAFS detection of the supernatant solution was performed.
References
Aragay, G., Pons, J. & Merkoci, A. Recent trends in macro-, micro- and nanomaterial-based tools and strategies for heavy-metal detection. Chem. Rev. 111, 3433–3458 (2011).
Orhan, A. Determination of cadmium, copper and lead in soils, sediments and sea water samples by ETAAS using a Sc + Pd + NH4NO3 chemical modifier. Talanta 65, 672–677 (2005).
Ashoka, S., Peake, B. M., Bremner, G., Hageman, K. J. & Reid, M. R. Comparison of digestion methods for ICP-MS determination of trace elements in fish tissues. Anal. Chim. Acta 653, 191–199 (2009).
Aranda, P. R., Pacheco, P. H., Olsina, R. A., Martinez, L. D. & Gil, R. A. Total and inorganic mercury determination in biodiesel by emulsion sample introduction and FI-CV-AFS after multivariate optimization. J. Anal. At. Spectrom. 24, 1441–1445 (2009).
Yuan, C. G., Wang, J. & Jin, Y. Ultrasensitive determination of mercury in human saliva by atomic fluorescence spectrometry based on solidified floating organic drop microextraction. Microchim Acta 177, 153–158 (2012).
Pourreza, N., Parham, H., Kiasat, A. R., Ghanemi, K. & Abdollahi, N. Solid phase extraction of mercury on sulfur loaded with N-(2-chlorobenzoyl)-N′-phenylthiourea as a new adsorbent and determination by cold vapor atomic absorption spectrometry. Talanta 78, 1293–1297 (2009).
Bagheri, H. & Naderi, M. Immersed single-drop microextractionelectrothermal vaporization atomic absorption spectroscopy for the trace determination of mercury in water samples. J. Hazard Mater. 165, 353–358 (2009).
Slaveykova, V. I. & Wilkinson, K. J. Physicochemical aspects of lead bioaccumulation by Chlorella vulgaris. Environ. Sci. Technol. 36, 969–975 (2002).
Mylon, S. E., Twining, B. S., Fisher, N. S. & Benoit, G. Relating the speciation of Cd, Cu and Pb in two Connecticut rivers with their uptake in algae. Environ. Sci. Technol. 37, 1261–1267 (2003).
Smolders, E. & McLaughlin, M. J. Chloride increases cadmium uptake in Swiss chard in a resin-buffered nutrient solution. Soil Sci. Soc. Am. J. 60, 1443–1447 (1996).
Kalis, E. J., Weng, L., Dousma, F., Temminghoff, E. J. & Van Riemsdijk, W. H. Measuring free metal ion concentrations in situ in natural waters using the Donnan membrane technique. Environ. Sci. Technol. 40, 955–961 (2006).
Clever, G. H., Kaul, C. & Carell, T. DNA–Metal Base Pairs. Angew. Chem., Int. Ed. 46, 6226–6236 (2007).
Willner, I. & Zayats, M. Electronic aptamer-based sensors. Angew. Chem., Int. Ed. 46, 6408–6418 (2007).
Tang, C. X., Zhao, Y., He, X. W. & Yin, X. B. A “turn-on” electrochemiluminescent biosensor for detecting Hg2+ at femtomole level based on the intercalation of Ru(phen)32+ into ds-DNA. Chem. Commun. 46, 9022–9024 (2010).
Ono, A. et al. Specific interactions between silver(i) ions and cytosine-cytosine pairs in DNA duplexes. Chem. Commun. 44, 4825–4827 (2008).
Xiao, Y., Rowe, A. A. & Plaxco, K. W. Electrochemical detection of parts-per-billion lead via an electrode-bound DNAzyme assembly. J. Am. Chem. Soc. 129, 262–263 (2007).
Li, T., Dong, S. & Wang, E. A Lead(II)-Driven DNA Molecular Device for Turn-On Fluorescence Detection of Lead(II) Ion with High Selectivity and Sensitivity. J. Am. Chem. Soc. 132, 13156–13157 (2010).
Vallée-Bélisle, A. & Plaxco, K. W. Structure-switching biosensors: inspired by Nature. Curr. Opin. Struct. Biol. 20, 1–9 (2010).
Marazuela, M. D. & Moreno-Bondi, M. C. Fiber-optic biosensors - an overview. Anal. Bioanal. Chem. 372, 664–682 (2002).
Mercury Update: Impact of Fish Advisories. EPA Fact Sheet EPA-823-F-01-011; EPA, Office of Water: Washington, DC, 2001.
Long, F., He, M., Zhu, A. N. & Shi, H. C. Portable optical immunosensor for highly sensitive detection of microcystin-LR in water samples. Biosens. Bioelectron. 24, 2346–2351 (2009).
ACS Committee on Environmental Improvement “Guidelines for Data Acquisition and Data Quality Evaluation in Environmental Chemistry”. Anal. Chem. 52, 2242–2249 (1980).
Wang, Z., Lee, J. H. & Lu, Y. Highly sensitive “turn-on” fluorescent sensor for Hg2+ in aqueous solution based on structure-switching DNA. Chem. Commun. 44, 6005–6007 (2008).
Nolan, E. M. & Lippard, S. J. Turn-on and ratiometric mercury sensing in water with a red-emitting Probe. J. Am. Chem. Soc. 129, 5910–5918 (2007).
Zhu, Z. et al. Highly sensitive electrochemical sensor for mercury(II) ions by using a mercury-specific oligonucleotide probe and gold nanoparticle-based amplification. Anal. Chem. 81, 7660–7666 (2009).
Wang, L. et al. Au NPs-enhanced surface plasmon resonance for sensitive detection of mercury(II) ions. Biosens. Bioelectron. 25, 2622–2626 (2010).
Yin, Y., Allen, H. E. & Huang, C. P. Kinetics of mercury(II) adsorption and desorption on soil. Environ. Sci. Technol. 31, 496–503 (1997).
Hoang, C. V., Oyama, M., Saito, O., Aono, M. & Nagao, T. Monitoring the presence of ionic mercury in environmental water by plasmon-enhanced infrared spectroscopy. Sci. Rep. 3, 1175; 10.1038/srep01175 (2013).
Long, F., He, M., Shi, H. C. & Zhu, A. N. Development of evanescent wave all-fiber immunosensor for environmental water analysis. Biosens. Bioelectron. 23, 952–958 (2008).
Method 245.7: Mercury in Water by Cold Vapor Atomic Fluorescence Spectrometry, U. S. Environmental Protection Agency Office of Water, Office of Science and Technology, Engineering and Analysis Division, February 2005 (http://water.epa.gov/scitech/methods/cwa/bioindicators/upload/2007_07_10_methods_method_245_7.pdf).
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (21077063, 21277173), the National Instrument Major Project of China (2012YQ3011105) and the Basic Research funds in Renmin University of China from the central government (13XNLJ01).
Author information
Authors and Affiliations
Contributions
F.L. and A.-N.Z. designed and performed all the experiments and wrote the manuscript. J.-Q.L. drew and summarized the figures. H.-C.S., H.-C.W. and J.-Q.L. finalized the preparation of the manuscript. F.L. designed and managed the project. All the authors discuss the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Long, F., Zhu, A., Shi, H. et al. Rapid on-site/in-situ detection of heavy metal ions in environmental water using a structure-switching DNA optical biosensor. Sci Rep 3, 2308 (2013). https://doi.org/10.1038/srep02308
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02308
This article is cited by
-
Recent advancement in development and modification of nanofibrous matrix for the application in sensing and remediation of water pollutants
Applied Nanoscience (2023)
-
Recent advancements in nanotechnological approaches for pollution monitoring and environmental sustainability
Clean Technologies and Environmental Policy (2023)
-
Recent Advances in Biosensors for Detection of Chemical Contaminants in Food — a Review
Food Analytical Methods (2022)
-
Concentration-dependent emission of nitrogen-doped carbon dots and its use in hazardous metal-ion detection
Carbon Letters (2021)
-
Quantum dot (QD)-based probes for multiplexed determination of heavy metal ions
Microchimica Acta (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
