
Abstract
London dispersion force is ubiquitous in nature and is increasingly recognized to be an important factor in a variety of surface processes. Here we demonstrate unambiguously the decisive role of London dispersion force in non-equilibrium growth of ordered nanostructures on metal substrates using aromatic source molecules. Our first-principles based multi-scale modeling shows that a drastic reduction in the growth temperature, from ~1000°C to ~300°C, can be achieved in graphene growth on Cu(111) when the typical carbon source of methane is replaced by benzene or p-Terphenyl. The London dispersion force enhances their adsorption energies by about (0.5–1.8) eV, thereby preventing their easy desorption, facilitating dehydrogenation and promoting graphene growth at much lower temperatures. These quantitative predictions are validated in our experimental tests, showing convincing demonstration of monolayer graphene growth using the p-Terphenyl source. The general trends established are also more broadly applicable in molecular synthesis of surface-based nanostructures.
Similar content being viewed by others
Introduction
London dispersion force1,2 describes the weak interaction between transient dipoles or multipoles associated with different parts of matter and is a major component of the more widely known van der Waals (vdW) force3. It is rooted in electron correlation effects, giving rise to long-range attractions between polar or non-polar materials. Because of the universal existence of transient dipoles1, London dispersion force is ubiquitous in nature, but its importance in various physical and chemical processes starts to be recognized only recently. Compelling examples include convincing demonstrations of the vital roles played by the vdW forces in surface migration4, organic/metal junction mechanics5 and molecular assembly at surfaces6,7. To a large extent, such advances through quantitative definitive studies were enabled by the availability of more accurate descriptions of the weak interactions associated with long-range electron correlation effects within first-principles approaches8,9,10.
In this study, we exploit the power of predictive modeling using state-of-the-art first-principles calculations within density functional theory (DFT), coupled with kinetic rate equation analysis and definitive experimental tests, to establish unambiguously the decisive role of London dispersion force in molecular self-assembly of aromatic source molecules. We choose graphene growth on Cu substrates as an important class of prototypical model systems and the microscopic mechanisms revealed are broadly applicable in other nanofabrication processes via molecular assembly.
Because of its exotic electronic properties11,12,13,14,15, graphene has been extensively studied as a candidate material with immense application potentials. Earlier developments mainly relied on graphene samples mechanically exfoliated using the “scotch-tape” approach16. More recently, increasing attention is paid to exploring alternative ways to achieve massive production of high-quality graphene, with graphene growth on metal substrates via chemical vapor deposition (CVD)17,18,19 as a prime example. In such experiments, the typical carbon source is methane (CH4) gas, requiring high growth temperatures of ~1000°C. Because CVD growth at such high temperatures is undesirable for many practical reasons, efforts have also been made to lower the growth temperature. For example, by using polymers as the carbon sources, graphene growth at the 800°C has been achieved20. More strikingly, it has been demonstrated that dramatic reductions in the growth temperature, from ~1000°C to (300–450)°C, could be achieved when different aromatic carbon sources were used in CVD growth21,22,23 of graphene on Cu or bottom-up fabrication24 of graphene nanoribbons on Au. Despite these apparent successes and highly desirable fabrication conditions, the underlying physical reasons remain to be explored and such insights are critical in future more precise design of graphene and other surface-based nanostructures.
Specifically, our DFT studies show that, whereas the inclusion of the vdW interaction has a minimal effect on the adsorption of CH4 on Cu(111), it dramatically enhances the bindings of benzene (C6H6) and p-Terphenyl (C18H14). Furthermore, using the DFT-based rate processes as input parameters, our detailed kinetic rate equation analysis reveals that the enhanced bindings help to prevent the molecules from easy desorption, and, more crucially, facilitate dehydrogenation and subsequent graphene growth at much lower temperatures. We also present preliminary experimental results that quantitatively validate the predictions for the case of C18H14.
Results
Enhanced bindings of the aromatic molecules
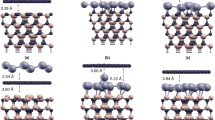
We first investigate the effects of London dispersion force on the adsorption of three different hydrocarbon molecules (CH4, C6H6 and C18H14) on Cu(111), whose connection with Cu foils will be discussed later. C6H6 and C18H14 are sp2-hybridized aromatic molecules (see Figs. 1b, c), which are expected to exhibit stronger London dispersion forces than CH4. To verify this expectation, we compare the results of DFT calculations using two different schemes, the vdW-DF8,10 and the Perdew-Burke-Ernzerhof (PBE)25 parametrization of the generalized gradient approximation. The optimized adsorption geometries from the vdW-DF calculations are shown in Fig. 1 (see also Supplementary Fig. S1) together with the corresponding adsorption energies (Eads) defined by


Atomic geometries and adsorption energies.
Top views of the optimized structures of (a) methane (CH4), (b) benzene (C6H6) and (c) p-Terphenyl (C18H14) on the Cu(111) surface, as obtained from the vdW-DF calculations. (d) The calculated adsorption energies of the three different molecules within the PBE and the vdW-DF schemes.
Here, the first, second and third term on the right hand side represents the total energy for the pure Cu substrate, the hydrocarbon molecule in gas phase and the combined adsorption system, respectively.
As shown in Fig. 1d, the adsorption energies within the vdW-DF scheme are 0.17, 0.67 and 1.93 eV for CH4, C6H6 and C18H14, respectively, showing a steep increase with the molecular size. For benzene, the value of Eads is close to the experimentally measured one of ~0.6 eV26 and is comparable to the value using a different vdW scheme27. Qualitatively, molecules of larger sizes are expected to exhibit more pronounced charge density fluctuations, caused by more delocalized motion of the electrons. Moreover, the more planar nature of the aromatic molecules also helps the carbon atoms to be located closer to the surface. Both of these factors help to enhance the attractive London dispersion forces. In contrast, our detailed PBE calculations yielded Eads to be 0.02, 0.07 and 0.11 eV for the three different hydrocarbon molecules, showing little variation in Eads in spite of the very different molecular sizes. Because the vdW-DF scheme was formulated to include the long-range correlation effects, while the semi-local PBE scheme is incapable of describing such effects8, the adsorption energy differences between the vdW-DF and the PBE schemes can be mainly attributed to the London dispersion force.
Energy barriers against dehydrogenation of the adsorbed molecules
The magnitude of the adsorption energy plays an important role in the competition between desorption and dehydrogenation since the adsorption energy is the same as the desorption barrier in the case of barrierless adsorption (see upper panel in Fig. 2). For example, an adsorbed molecule is most likely to desorb from the surface back to the gas phase than to dehydrogenate if its adsorption energy is much less than the potential energy barrier (Eb) against dehydrogenation. For further quantitative investigations, we have calculated Eb using the nudged elastic band method28 for different adsorbed molecules. In these calculations, we consider only the vdW-DF scheme because the PBE method provides a poor description of the adsorption energetics of the molecule/metal systems6,29. The calculated energy profiles are shown in Fig. 2. We find that Eb is 1.53, 1.51 and 1.55 eV for CH4, C6H6 and C18H14, respectively, exhibiting little difference from one type of molecules to another, an important finding different from an earlier study21 without consideration of the vdW interaction. Most significantly, the enhanced bindings will facilitate dehydrogenation of the adsorbed C6H6 or C18H14 molecules by prohibiting their easy desorption, especially for the latter, which have a lower dehydrogenation barrier than desorption. In contrast, it is most difficult for the adsorbed CH4 molecules to dehydrogenate, because their desorption barrier of 0.17 eV is one order of magnitude smaller than the dehydrogenation barrier of 1.53 eV. The calculated Boltzmann factor for the energy difference of 1.36 eV and temperature of 300°C is ~10−12, indicating almost zero probability of dehydrogenation for an adsorbed methane molecule. Therefore, graphene growth using CH4 as the carbon source can only proceed at much higher temperatures and supersaturated gas pressures.

Calculated energetics and kinetics for the adsorption and dehydrogenation of the three different kinds of molecules on the Cu(111) surface.
The atomic geometries of the adsorption, transition and dehydrogenated states are also given for each type of molecules. The abbreviation of GS, AS, TS and DS represents the gaseous, adsorbed, transition and dehydrogenated state, respectively. The reference energy is the total energy of the system before adsorption, calculated as Etot(Cu) + Etot(molecule).
Estimation of the growth temperatures via kinetic rate equation analysis
Now we quantify the time scales for desorption and dehydrogenation based on the energetics and kinetics presented in Fig. 2, so as to estimate the graphene growth temperatures determined by the delicate competitions of the atomistic rate processes. First, we note that under equilibrium conditions, CH4 or C6H6 molecules adsorbed on the surface will more likely desorb than dehydrogenate, while C18H14 will more likely dehydrogenate than desorb. Nevertheless, it has been shown experimentally that the use of CH4 or C6H6 as carbon sources does lead to graphene growth. In such cases, growth was achieved under non-equilibrium growth conditions, where a finite gas pressure establishes supersaturation of the molecules in the gas phase and the growth temperature is sufficiently high to overcome the dehydrogenation barrier(s). The number (ND) of the desorbed molecules during one dehydrogenation event can be estimated quantitatively by the ratio of the desorption rate (Rdes) over the dehydrogenation rate (Rdeh). Therefore, the time needed for a dehydrogenation event, tD, is the same as the time needed for ND desorption events. The latter can be estimated by the physically reasonable assumption that the desorption reactions occur as frequently as min[Rads, Rdes], namely, the smaller between the adsorption rate (Rads) and the desorption rate. In contrast to CH4 and C6H6, the desorption rate for C18H14 will be much slower than the dehydrogenation rate. Therefore, for C18H14, desorption can be ignored and tD is obtained directly from the dehydrogenation rate. The Rads, Rdes and Rdeh values based on our DFT results are estimated by following an existing approach30 that combined DFT calculations with transition state theory (TST). The estimated values are given in Supplementary Table S1. From the estimated reaction rates, we obtain the expected dehydrogenation time tD at different temperatures as shown in Table 1.
Table 1 shows an increase in tD with decreasing temperature for each type of molecules. This is mainly because the dehydrogenation rate is proportional to exp(−Eb/KBT), where KB is the Boltzmann constant and T is the temperature. It is particularly worthwhile to note that, even though the dehydrogenation barrier Eb is essentially identical for all the three types of molecules, the effective time tD needed for one dehydrogenation event is distinctively different as a result of the delicate competitions between the various kinetic rate processes. Furthermore, the Rads values of CH4 and C6H6 depend sensitively on the molecular pressure, making the corresponding tD and consequently the growth rate of graphene to be also gas-pressure dependent. In contrast, since the desorption rate of C18H14 is negligible, graphene growth can proceed at even lower gas pressures, as long as the growth temperature is sufficiently high.
Since sustaining graphene growth on Cu requires complicated multi-step kinetic processes of adsorption, dehydrogenation, carbon coalescence and other atomistic/molecular events, the actual value of tD in a real non-equilibrium growth system should be substantially shorter than the experimental growth time adopted. The typical experimental graphene growth time is less than one hour, requiring that tD be much smaller than one hour. Based on these considerations and the results in Table 1, we conclude that graphene growth can proceed at 300°C if using C6H6 as the carbon source, as previously observed experimentally21. In contrast, the tD values for CH4 below 400°C are much larger than the typical experimental graphene growth time, indicating that graphene growth using CH4 as the carbon source must take place at much higher temperatures. This observation is again consistent with existing experiments17,21. It is noticeable that C18H14 also has a short tD at 300°C, only 3.6 seconds, implying that graphene growth using the C18H14 source is possible at this temperature. At the even lower temperature of 200°C, tD for both C6H6 and C18H14 is comparable to the typical experimental growth time, thereby ruling out the possibility of growing graphene at such a low temperature. Based on these discussions we can define a temperature boundary for each type of molecules, marked by the symbols √ and × in the parentheses in Table 1; this boundary separates the temperature range for success (above) or failure (below) to experimentally achieve graphene growth. Table 1 clearly shows that the dehydrogenation is the rate-limiting process of the graphene growth, defining the lower boundary of growth temperature. After the dehydrogenation, the molecules must diffuse on the substrate to make C-C connections with other molecules or nucleated graphene flake. The calculated potential energy barrier against diffusion is 0.08, 0.22 and 0.34 eV for the dehydrogenated methane, benzene and p-Terphenyl, respectively. Therefore, the dehydrogenated molecules can easily diffuse on the substrate and effectively form the C-C connections. To further reduce the growth temperature, it is desirable to devise new catalytic processes to reduce the potential energy barrier against dehydrogenation.
Validation of the predictions via definitive experimental tests
We now make closer connections with existing experimental results and new growth experiments designed to test the strong predictions made above. As reported in a previous study21, no graphene growth was observed on Cu foils from the methane source when the growth temperature was lowered to 600°C. In contrast, when benzene was used as the feedstock, growth of monolayer graphene flakes with excellent quality was achieved at the drastic reduced temperature of 300°C21, as shown in the scanning electron microscope (SEM) image and the Raman spectrum (Fig. 3a). As a further validation of the theoretical results presented above, here we show results of graphene growth using p-Terphenyl as the carbon source at 300°C, exhibiting the continuous monolayer graphene films grown under these conditions (Figs. 3b–3d). First, in both inserts of Figs. 3a and 3b, the Raman spectra reveal the typical characteristics of monolayer graphene: The 2D bands around the ~2685 cm−1 are symmetric and can be well fitted by a single Lorentzian distribution31,32. For either case, the full width at half maximum (FWHM) of the 2D band is ~35 cm−1 and the intensity ratio of the G band to the 2D band is ~0.5. These features are very similar to those for CH4-derived monolayer graphene17. The low-intensity D band (~1348 cm−1) in Fig. 3b is likely derived from the more populous domain boundaries shown in the SEM image. Comparing the Raman spectra to those of amorphous carbon in the previous experiment33, it is noticeable that the C-C bonds in the grown graphene are primarily sp2 in nature without considerable signal of sp3 bonds. This is because the CVD growth of graphene on Cu substrates relies on the catalytic function of Cu to dehydrogenate the molecules, resulting in graphene growth of predominently monolayer in thickness. Therefore, second layer growth of graphene rarely occurs, no superstructures over the monolayer graphene and similarly, formation of amorphous carbon can be suppressed. Secondly, the optical transmittance at 550 nm for the p-Terphenyl derived graphene grown at 300°C was measured to be 96.8% (Fig. 3c), which is close to the value of 97.1% reported for monolayer graphene20. The Raman and optical transmittance data collectively establish the formation of monolayer graphene at macroscopic sizes. Finally, the scanning tunneling microscopy (STM) characterization has also been conducted on the as-grown graphene on the copper foils and the results are shown in Fig. 3d. The atomic-scale STM imaging of the graphene clearly reveals its honeycomb pattern, which has been well established to characterize its monolayer nature18,34,35. Together, these experimental characterizations convincingly establish that the graphene films grown using the p-Terphenyl source at 300°C are predominantly monolayer in thickness. We have also tried to grow graphene at a lower temperature of 200°C using benzene or p-Terphenyl as the carbon source, but achieved no graphene growth, because the corresponding tD would be too long (see the last row in Table 1). These experimental observations are in excellent quantitative agreement with the theoretical predictions and collectively, these results unambiguously establish the decisive role of the London dispersion force in low-temperature growth of graphene using the aromatic carbon sources.

Characterization of the graphene films grown at 300°C from different carbon sources.
(a) and (b) SEM images of the graphene films derived from benzene and p-Terphenyl, respectively. The corresponding Raman spectra are also given in the insets. (c) Optical transmittance spectrum of the graphene film using the p-Terphenyl source, with that at 550 nm indicated by the dashed line. (d) STM images of the graphene film using the p-Terphenyl source at the sample bias of −0.1 V, with the insert highlighting honeycomb structure.
Discussion
Before closing, we note that the Cu foils used in typical CVD graphene growth were polycrystalline in structure36. Similarly, the Cu foils used in our present experiments also contain other facet orientations such as (100) and (110), rather than purely (111). However, it has been established experimentally that the Cu(111) surface is able to catalyze the highest quality of graphene growth with larger sizes and shorter growth times than other facets; therefore, it is more desirable to produce higher densities of Cu(111) facets in the Cu foils36. On the other hand, London dispersion force is an universal interaction and therefore it can play a similar role in the graphene CVD growth on other Cu facets. This intriguing aspect will be investigated in future control experiments, where graphene growth on different surfaces of crystalline Cu will be investigated using different hydrocarbon source molecules.
At this point, it is quite natural to emphasize that the general trends established above should also be operative in CVD growth of graphene employing other aromatic carbon sources. One such example was the demonstration of CVD graphene growth on Cu at (300–360)°C using pyridine (C5H5N) or hexachlorobenzene (C6Cl6) as the carbon source22,23; another striking example was the bottom-up fabrication of ordered graphene nanoribbons on Au at ~450°C or lower using more complex but inherently aromatic carbon sources24. In both cases, the London dispersion forces must have played a crucial role in enabling low-temperature growth of graphene or graphene nanostructures.
Aside from graphene growth using different aromatic source molecules discussed above22,23,24, here we also briefly discuss the potential broader applicability of the main findings in the fabrication of other nanostructures catalyzed on transition metal substrates as reported recently37,38,39. A closer comparison between these seemingly very different systems leads to the recognition of several essential conceptual similarities. First, all these (the previous and present) studies used carbon-based aromatic source molecules, though those molecules differ in sizes and specific chemical modifications. Secondly, in fabricating the desired nanostructures as ending products, certain chemical processes have to take place and in several of the examples the specific chemical processes included dehydrogenation of the source molecules and subsequent formation of C-C bonds. Thirdly, the chemical processes need to be efficiently facilitated with proper choices of the catalytic substrates and ideally taking place at low or moderate substrate temperatures. Finally, the ending products (graphene or fullerenes or others) possess much stronger structural stabilities than the related nanostructures consisting of the aromatic source molecules weakly bounded together via the London dispersion forces, due primarily to the occurrence of the chemical processes. Given these conceptual similarities, it is highly desirable to revisit those earlier studies, to see how critical it is to include the London dispersion forces in understanding quantitatively the various adsorption and subsequent chemical reaction processes of the different aromatic source molecules. The ubiquitous nature of the London dispersion force ensures that its importance is bound to be increasingly exploited in future studies of various surface kinetic, dynamic and catalytic processes of technological significance.
Methods
DFT calculations
The DFT calculations were performed with the Vienna Ab-initio Simulation Package (VASP) simulation package, employing a 400-eV plane-wave cutoff and projector augmented-wave (PAW)40 potentials. The Cu(111) surface was simulated by a five-layer p(6 × 6) unit cell containing 180 Cu atoms. The thickness of the vacuum region was set to ~17 Å. In the p(6 × 6) supercell, the intermolecular distance of methane is much longer than that of the two aromatic molecules. Therefore, we have checked the adsorption energies of methane with smaller supercells down to p(2 × 2) Cu(111). However, the adsorption energy increases by only ~0.02 eV, indicating weak intermolecular London dispersion interactions even for the highest densities of methane considered. The calculated Cu equilibrium lattice constants are 3.6258 Å and 3.5978 Å within the PBE and vdW-DF schemes, respectively and both values agree well with that of experiment (3.61 Å). The surface Brillouin zone was sampled using a 2 × 2 × 1 Monkhorst-Pack k-point mesh. All the atoms except the bottom two Cu layers were allowed to relax, while the bottom two layers were fixed in their respective bulk positions.
Reaction rate analysis
The adsorption rate Rads is defined by the number of events per unit time onto an adsorption site of area Aas. At temperature T and molecular pressure p, it can be obtained by the following equation30,

where m is the mass of a molecule and KB is the Boltzmann constant. Here, we set the sticking coefficient S(T) to unity, assuming barrierless adsorption and no further reduction in the sticking probability. The molecular pressure was taken to be 10 torr, which is in the same range of the experimental values. We also assumed Aas as the area of p(1 × 1), p(2 × 2) and three times of p(2 × 2) Cu(111) surface for CH4, C6H6 and C18H14 adsorptions, respectively. The values of Rdes and Rdeh were estimated by the following TST form30:

where Ea is the activation energy and the prefactor A is KBT/h30. Here, h is the Planck constant.
Graphene growth from benzene source
Cu foils were initially cleaned at 1000°C in a 100 sccm H2 flow for 20 min without the benzene source in the quartz tube and then cooled to room temperature. After the pretreatment, liquid benzene loaded in a small glass container was placed at the gas inlet side of the quartz tube, just outside of the heating zone. Then the furnace was heated to the desired growth temperatures. The growth time was usually between 15–30 min, with a 50 sccm H2 flow during growth while maintaining the total pressure to be within (8–15) Torr. After growth, the synthesized graphene was transferred onto SiO2/Si substrates for Raman and SEM characterizations, as well as onto quartz substrates for optical transmittance measurements.
Graphene growth from p-Terphenyl source
The solid p-Terphenyl molecules loaded in a small glass container were placed at the gas inlet side of the quartz tube, just outside of the heating zone. Independent heating control for the solid sources was supplied by a heating tape. Before growth, Cu foils were cleaned at 1000°C in a 100 sccm H2 flow for 20 min and then cooled down to the desired growth temperatures. The solid precursors were then heated to about 200°C by the heating tape with the H2 flow reduced to 50 sccm while maintaining the total pressure to be within (8–15) Torr. The typical growth time was about 45 min. After growth, the furnace was opened for fast cooling. The as-grown graphene films were then transferred onto SiO2/Si substrates for Raman and SEM characterizations, as well as onto quartz substrates for optical transmittance measurements.
Sample characterizations
Raman spectroscopy (French JY LABRAM-HR) with laser excitation wavelength of 514.5 nm was used to characterize the thickness, quality and uniformity of the grown graphene films at room temperature. The morphology of the graphene films was characterized using a field emission SEM (FEI Sirion 200) operated at 5 kV. Optical transmittance spectra were measured using a UV-VIS-NIR spectrophotometers (Shimadzu DUV-3700). The STM characterization was performed on the as-grown graphene films on Cu foils at 78 K using a Createc low-temperature STM.
References
London, F. The General Theory of Molecular Forces. Trans. Faraday Soc. 33, 8–26 (1937).
Stone, A. J. The Theory of Intermolecular Forces (Oxford University Press, Oxford, 1997).
Van der Waals, J. D. The thermodynamic theory capillarity under the hypothesis of a continuous variation of density. Verhandel. Konink. Akad. Weten. Amsterdam 1, 8 (1893); translation published by Rowlinson J. S. J. Stat. Phys. 20, 197–200 (1979).
Zhang, Y. N., Hanke, F., Bortolani, V., Persson, M. & Wu, R. Q. Why sliding friction of Ne and Kr monolayers is so different on the Pb(111) surface. Phys. Rev. Lett. 106, 236103 (2011).
Aradhya, S. V., Frei, M., Hybertsen, M. S. & Venkataraman, L. Van der Waals interactions at metal/organic interfaces at the single–molecule level. Nat. Mater. 11, 872–876 (2012).
Bartels, L. Tailoring molecular layers at metal surfaces. Nat. Chem. 2, 87–95 (2010).
Björk, J., Stafström, S. & Hanke, F. Zipping up: cooperativity drives the synthesis of graphene nanoribbons. J. Am. Chem. Soc. 133, 14884–14887 (2011).
Dion, M., Rydberg, H., Schröder, E., Langreth, D. C. & Lundqvist, B. I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 92, 246401 (2004).
Tkatchenko, A. & Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 073005 (2009).
Román-Pérez, G. & Soler, J. M. Efficient implementation of a van der Waals density functional: application to double-wall carbon nanotubes. Phys. Rev. Lett. 103, 096102 (2009).
Novoselov, K. S. et al. Two-dimensional gas of massless Dirac fermions in graphene. Nature 438, 197–200 (2005).
Zhang, Y. B., Tan, Y. W., Stormer, H. L. & Kim, P. Experimental observation of the quantum Hall effect and Berry's phase in graphene. Nature 438, 201–204 (2005).
Berger, C. et al. Electronic confinement and coherence in patterned epitaxial graphene. Science 312, 1191–1196 (2006).
Bostwick, A., Ohta, T., Seyller, T., Horn, K. & Rotenberg, E. Quasiparticle dynamics in graphene. Nat. Phys. 3, 36–40 (2007).
Castro Neto, A. H., Guinea, F., Peres, N. M. R., Novoselov, K. S. & Geim, A. K. The electronic properties of graphene. Rev. Mod. Phys. 81, 109–162 (2009).
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Li, X. S. et al. Large-area synthesis of high-quality and uniform graphene films on copper foils. Science 324, 1312–1314 (2009).
Gao, L., Guest, J. R. & Guisinger, N. P. Epitaxial graphene on Cu(111). Nano Lett. 10, 3512–3516 (2010).
Bhaviripudi, S., Jia, X., Dresselhaus, M. S. & Kong, J. Role of kinetic factors in chemical vapor deposition synthesis of uniform large area graphene using copper catalyst. Nano Lett. 10, 4128–4133 (2010).
Sun, Z. Z. et al. Growth of graphene from solid carbon sources. Nature 468, 549–552 (2010).
Li, Z. C. et al. Low-temperature growth of graphene by chemical vapor deposition using solid and liquid carbon sources. ACS Nano 5, 3385–3390 (2011).
Gan, X. C. et al. A simple method to synthesize graphene at 633 K by dechlorination of hexachlorobenzene on Cu foils. Carbon 50, 306–310 (2012).
Xue, Y. Z. et al. Low temperature growth of highly nitrogen-doped single crystal graphene arrays by chemical vapor deposition. J. Am. Chem. Soc. 134, 11060–11063 (2012).
Cai, J. M. et al. Atomically precise bottom-up fabrication of graphene nanoribbons. Nature 466, 470–473 (2010).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Lukas, S., Vollmer, S., Witte, G. & Wöll, Ch. Adsorption of acenes on flat and vicinal Cu(111) surfaces: step induced formatioin of lateral order. J. Chem. Phys. 114, 10123–10130 (2001).
Ruiz, V. G., Liu, W., Zojer, E., Scheffler, M. & Tkatchenko, A. Density-functional theory with screened van der Waals interactions for the modeling of hybrid inorganic-organic systems. Phys. Rev. Lett. 108, 146103 (2012).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Li, G., Tamblyn, I., Cooper, V. R., Gao, H.-J. & Neaton, J. B. Molecular adsorption on metal surfaces with van der Waals density functionals. Phys. Rev. B 85, 121409(R) (2012).
Reuter, K. & Scheffler, M. First-principles kinetic Monte Carlo simulations for heterogeneous catalysis: application to the CO oxidation at RuO2(110). Phys. Rev. B 73, 045433 (2006).
Ferrari, A. C. et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 97, 187401 (2006).
Graf, D. et al. Spatially resolved Raman spectroscopy of single- and few-layer graphene. Nano Lett. 7, 238–242 (2007).
Ferrari, A. C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 143, 47–57 (2007).
Yu, Q. et al. Control and characterization of individual grains and grain boundaries in graphene grown by chemical vapour deposition. Nat. Mater. 10, 443–449 (2011).
Stolyarova, E. et al. High-resolution scanning tunneling microscopy imaging of mesoscopic graphene sheets on an insulating surface. Proc. Natl. Acad. Sci. 104, 9209–9212 (2007).
Wood, J. D., Schmucker, S. W., Lyons, A. S., Pop, E. & Lyding, J. W. Effects of polycrystalline Cu substrate on graphene growth by chemical vapor deposition. Nano Lett. 11, 4547–4554 (2011).
Otero, G. et al. Fullerenes from aromatic precursors by surface-catalysed cyclodehydrogenation. Nature 454, 865–869 (2008).
Gourdon, A. On-surface covalent coupling in ultrahigh vacuum. Angew. Chem. Int. Ed. 47, 6950–6953 (2008).
Rim, K. T. et al. Forming aromatic hemispheres on transition-metal surfaces. Angew. Chem. Int. Ed. 46, 7891–7895 (2007).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Acknowledgements
We thank Guo Li for helpful discussions. This work was supported in part by the Chinese Academy of Sciences Fellowships for Young International Scientists (J.-H.C., 2011Y2JB10), NSFC (11034006, 91021018, 11250110056 and 11204286), FRFCU (WK2340000011 and WK2340000029), “One-hundred-person Project” of CAS, NKBRPC (2009CB929502), USNSF (0906025), USDOE (ER45958) and NRF of Korea (2012R1A6A3A03040199).
Author information
Authors and Affiliations
Contributions
J.-H.C. and Z.Z. conceived the idea for the theories and wrote the manuscript. J.-H.C. and P.C. performed the density functional theory calculations. C.Z. designed the experiments and co-wrote the manuscript. Z.L., X.F. and H.Z. prepared and characterized the samples. All authors discussed the results and edited the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Choi, JH., Li, Z., Cui, P. et al. Drastic reduction in the growth temperature of graphene on copper via enhanced London dispersion force. Sci Rep 3, 1925 (2013). https://doi.org/10.1038/srep01925
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01925
This article is cited by
-
Controlled CVD preparation and quality characterization of graphene based on biphenyl refined from low-rank coal
Carbon Letters (2023)
-
Disorder-tuned conductivity in amorphous monolayer carbon
Nature (2023)
-
Synthesis of Large-Area Single-Layer Graphene Using Refined Cooking Palm Oil on Copper Substrate by Spray Injector-Assisted CVD
Nanoscale Research Letters (2019)
-
Recent trends in graphene materials synthesized by CVD with various carbon precursors
Journal of Materials Science (2018)
-
Near room temperature chemical vapor deposition of graphene with diluted methane and molten gallium catalyst
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
