
Abstract
In the Red Sea, two neighboring deep-sea brine pools, Atlantis II and Discovery, have been studied extensively and the results have shown that the temperature and concentrations of metal and methane in Atlantis II have increased over the past decades. Therefore, we investigated changes in the microbial community and metabolic pathways. Here, we compared the metagenomes of the two pools to each other and to those of deep-sea water samples. Archaea were generally absent in the Atlantis II metagenome; Bacteria in the metagenome were typically heterotrophic and depended on aromatic compounds and other extracellular organic carbon compounds as indicated by enrichment of the related metabolic pathways. In contrast, autotrophic Archaea capable of CO2 fixation and methane oxidation were identified in Discovery but not in Atlantis II. Our results suggest that hydrothermal conditions and metal precipitation in the Atlantis II pool have resulted in elimination of the autotrophic community and methanogens.
Similar content being viewed by others

Introduction
Along the narrow seafloor of the Red Sea, approximately 25 deep-sea brine pools have been formed by the spreading of the Arabic and African plates1. The movement of these plates results in volcanic activity due to breaking of the earth crust. Injection of a geothermal solution into seafloor depression resulted in the development of deep-sea brine pools due to mixing of the hot and metalliferous solution with seawater2,3. Two famous brine pools, Atlantis II and Discovery, were discovered in the 1960s and since then, these brine pools have been the focus of geological and geochemical surveys in the Red Sea4,5,6,7. A number of geological questions have been raised, including questions about the relationship between the two brine pools. Because of their close proximity, it has been suggested that the Atlantis II and Discovery are connected and that brine flows between the two pools8. This hypothesis was supported by a parallel change in the anhydrite content in the sediment pore water9. Given their proposed historic connection and similar environments, it has been suggested that the pools were inhabited by similar microbial communities that can be exchanged in between.
It is now known that the two brine pools are separated by a sill, which is about 50 m higher than the main brine levels10 and the two pools have different environmental conditions. The most striking observation was a gradual increase in temperature over the last few decades in the Atlantis II pool from 56 to 68°C11,12, while the Discovery brine pool has maintained a stable temperature of approximately 44°C12. In addition, the methane concentration in the Atlantis II pool is 4-fold higher than that in the Discovery pool13. The concentrations of certain metal ions, such as Fe, Mn, Li and Zn, are also much higher in Atlantis II1. All of these differences have been attributed to mounting volcanic activity at the bottom of the Atlantis II basin; this activity has supplied the pool with a hydrothermal solution with estimated temperatures and salinities ranging from 195–310°C and 270–370‰, respectively14. The Atlantis II brine pool now has three upper convective layers that differ in salinity, temperature and metal content7. In contrast, only one upper layer is evident in the Discovery brine pool7. Therefore, since the two pools have different physical and geochemical conditions, it is hypothesized that the microbial inhabitants of these brine pools are also different and have different metabolic activities. The objective of this study was to test this hypothesis.
Our recent metagenomic work has provided a glimpse of the microbes that are present in the pools and focused on the organic maturation process in the Atlantis II pool15. However, a comprehensive comparison of the two metagenomes of the microbial communities and their activities has not been conducted. In addition the roles of the microbes in the carbon cycle and metal precipitation in the two brine pools have not been examined. Analyses of gene profiles and biodiversities within the pools could provide insight into the microbial fitness model in these extreme environments.
In the present study, we analyzed metagenomes from the lower convective layers in the Atlantis II and Discovery brine pools and compared them with two deep-sea water metagenomes. Strong dissimilarities between the brine pool metagenomes were demonstrated with respect to the microbial community and metabolic spectrum. The Atlantis II metagenome was enriched with heterotrophic metal-reducing microbes utilizing aromatic compounds and extracellular organic carbon sources, whereas autotrophic microbes capable of CO2 fixation and methane oxidation were found in Discovery and functioned as primary producers. Compared with the deep-sea water samples, the brine pool microbes displayed weaknesses in the glyoxylate cycle, oxidative phosphorylation and biosynthesis under anaerobic conditions.
Results
Different microbial communities in the two brine pools
Sampling sites in the Atlantis II and Discovery brine pools are shown in Figure 116. The temperature in the lower convective layer (>2100 m) was 68°C in Atlantis II and 45°C in Discovery. The salinity was 255‰ in both pools. The bacterial cell density in the Atlantis II and Discovery pools was 9,200 and 7,000 cells ml−3, respectively. The brine water samples were pyrosequenced (hereafter referred to as ‘ABP’ for Atlantis II; ‘DBP’ for Discovery). The 16S rRNA gene fragments were extracted from the brine metagenomes and two reference deep-sea water metagenomes (CAM: Carmen Basin in the Gulf of California and MED: the Matapan-Vavylov deep in the Mediterranean Sea) to obtain an initial overview of the microbial community based on the RDP classifier17 (Table 1). In CAM and DBP, Archaea occupied approximately 8% and 47% of their 16S rRNA fragments, respectively. The deep-sea water samples (CAM and MED) were dominated by Gammaproteobacteria as previously reported18,19, but this dominance was replaced by Betaproteobacteria in ABP (66%) and by Euryarchaeota in DBP (42%). Betaproteobacteria ranked second in DBP (28%). At the genus level, Cupriavidus (59% in ABP; 25% in DBP) was the major component of communities in the brine metagenomes (ABP and DBP) and Alteromonas (86%) dominated overwhelmingly the MED sample. Additional details about all the other genera have been previously described15,18,19. The percentage of known taxa below the phylum level dropped sharply in CAM and therefore, the dominant genera could not be determined. Approximately 30% of the 16S rRNA gene fragments of CAM could not be determined, consistent with the results of a metatranscriptomic study of a sample from a neighboring site20. At the order level, only 25% of the CAM fragments could be recognized; they were particularly difficult to sort into known taxonomies at lower levels, potentially due to the shorter reads obtained for CAM (Table 1).

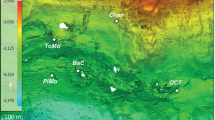
Geographic map of the sampling sites.
Map showing the two sample sites. The Atlantis II (ABP) and Discovery (DBP) brine pools in the Red Sea (inlet), where brine water was collected (at a depth >2100 m) using Niskin bottles. The map was based on bathymetric data obtained using the SIMRAD EK60 echosounder (Kongsberg Maritime AS, Noway) during the KAUST Red Sea Expedition 2010 and contoured using ArcGIS software (ESRI, USA).
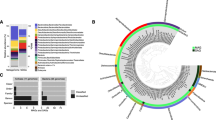
The taxonomy of a metagenome can also be examined based on gene content. Among the reads, 627,433 and 618,510 complete open reading frames (cORFs) were collected for ABP and DBP, respectively. Using the BLAST result for the curated cORFs, the taxonomies were summarized for ABP and DBP using MEGAN21 (Fig. 2). Consistent with the results based on the 16S rRNA gene fragments, Betaproteobacteria (70%) and Euryarchaeota (42%) were the dominant phyla in the corresponding brine microbial communities. Archaea accounted for approximately 50% of the whole community in DBP. In contrast, there were several archaeal reads in ABP. The Euryarchaeota in DBP was mainly composed of Archaeoglobales, Halobacteriales and a group of orders involved in methanogenesis and the anaerobic oxidation of methane (AOM). Regarding the bacterial phyla, DBP had a higher proportion of Firmicutes, Deinococcus-Thermus and Delta/Epsilonproteobacteria compared with ABP. Overall, DBP displayed a higher biodiversity than ABP, as suggested by its two-fold higher Shannon index (2.19 vs. 1.04).

Taxonomic assignment of ABP and DBP cORFs.
Taxonomic assignment was based on the taxa of the ABP and DBP cORFs following a BLAST search against the NCBI protein database. Archaea accounted for approximately 50% of the microbial community in DBP and they were then classified down to the level of order.
Categories of Clusters of Orthologous Groups (COG) in the four metagenomes
COGs in the reads were sorted into categories. The ABP and DBP brine samples did not consistently display an over or under-representation in the individual categories, as compared with the deep-sea samples (Fig. S1). ABP had the highest percentage of COGs in the following categories: carbohydrate transport and metabolism, lipid metabolism, transcription, intracellular trafficking and secretion and secondary metabolite biosynthesis, DBP featured the highest level of categories representing DNA replication, recombination and repair among the four metagenomes. This was in accord with the presence of abundant phages in DBP, as indicated by taxonomic assignment of the genes.
Examination of the Kyoto Encyclopedia of Genes and Genomes (KEGG) genes and pathways to show the relationship between samples
Because the results for the COG categories revealed a lack of congruency among the brine or the deep-sea samples, KEGG genes were used to examine differences and similarities among the samples. A Bray-Curtis dissimilarity matrix was calculated using standardized frequencies of the KEGG genes. Subsequently, a relationship based on an agglomerative hierarchical clustering (AHC) was displayed (Fig. 3). The position at which MED and CAM merged was below the primary merging dissimilarity level of 0.52 in AHC and therefore, they could be grouped together. Although ABP and DBP were obtained from adjacent brine pools, they could not be merged together. Principal coordinates analysis (PCoA) was employed to further illustrate the relationship between the samples. In a plot of the principal coordinates F1 and F2, the four samples were widely separated (Fig. 4), suggesting a distant relationship among the samples on a large scale. Despite this finding, close relationships were illustrated in pair-wise plots of other coordinates. ABP and CAM showed a closer relationship in the plot of F1 and F3; DBP and MED were the closest neighboring points in the plot of F2 and F3 (Fig. 4), which indicated that similarities in gene abundance persisted in a small fraction of the genes in DBP and MED. However, the results also indicated that environmental differences might have drastically shifted the microbial communities and their metabolic processes. Thus, we focused next on determining which genes and pathways were responsible for the disparities.

Agglomerative hierarchical clustering of KEGG genes.
The dashed line refers to the minimum dissimilarity level at which the samples were grouped.

Principal coordinate analysis results.
Principal coordinate analysis and the scree plot revealed three principal coordinates (axes). The three plots were generated using different combinations of the principal coordinates.
The KEGG genes were clustered based on their relative abundance after normalization according to the z-score. The results from the sample clustering using a complete linkage method were consistent with those obtained for the AHC analysis (Figs. 3 and 5). In the heat map of the gene profiles, sample-specific enriched gene clusters occupied a large part of the region, but commonly enriched clusters were rare, in agreement with the large distance between the samples in the plot of principal coordinates F1 and F2 (Figs. 4 and 5). There were pathways represented by the genes present in five hot regions, areas I to V in Figure 5. A high level of pathway completeness suggested their adaptive importance within a given sampling site. For instance, ABP-specific region I comprised genes affiliated with pathways that participate in the degradation of aromatic compounds and non-aromatic cyclic organic carbon, butanoate metabolism, nitrogen fixation, unsaturated fatty acids synthesis and lipid metabolism. The completeness of these pathways was striking. Together with the findings from the COG category analysis regarding carbohydrate transport and metabolism, the microbes present in ABP were generally heterotrophic bacteria that depended heavily on extracellular aromatic and other organic carbon sources.

Sorting of sample-specific enriched gene clusters into pathways.
The frequencies of KEGG genes were normalized using the z-score and clustered together with the samples. Genes present in the five regions of the heat map (I-V) were assigned to different KEGG pathways. The same pathways appeared in different regions, but the list of genes (sometimes encoding subunits of an enzyme) was different. The number of identified genes and the total number of enzymes in the pathways are indicated in parentheses.
Some of the gene clusters specific to DBP in region II were affiliated with CO2 fixation and methane metabolism and these pathways displayed a high level of completeness. Sugar utilization in DBP was inferred from the pathway map for starch and sucrose metabolism, in which starch, sucrose, isomaltose, extracellular trehalose and cellulose were potential sugar sources based on the Enzyme Commission (EC) numbers in the map. Region III for MED contained dozens of genes involved in carotenoid biosynthesis, nitrogen metabolism (denitrification), flagellar assembly and bacterial chemotaxis. Region V comprised genes that were commonly enriched in CAM and MED and showed a high completeness of the terpenoid biosynthesis pathway. For CAM and MED, regions III-V showed a strong representation of oxidative phosphorylation and cell membrane synthesis pathways among the enriched genes. The specific genes involved in oxidative phosphorylation differed among the three regions as discussed below.
Functional modules of the KEGG genes in Figure 5 are summarized in Table S1. Based on the completeness of the modules, metabolic and adaptive activities were demonstrated, particularly those conducted by ATP-binding cassette (ABC) transporters and two-component systems in a metagenome. Many transporter genes involved in the transmembrane transport of various amino acids and sugars were found in region I for ABP. Strikingly, almost all of the gene members in the type IV and VI secretion systems were enriched in ABP. A special assignment of genes into modules for V-type ATP generation (typically the archaeal type), methanogenesis and CO2 fixation highlighted their importance in DBP. Metal ion pumps of the cobalt/nickel/iron complex and manganese/iron showed fairly complete representation as well. In region III, the presence of cytochrome c oxidase genes in the cbb3 type was associated with a strong pentose phosphate module, which was indicative of a response to the low oxygen concentration in the MED sampling site19,22. Genes encoding ABC polysaccharide transporters were also concentrated in this region. In addition, the type II general secretion system was highly represented in MED. For CAM, region IV was characterized by genes specific to pathways that participate in NADH dehydrogenation (complex I in oxidative phosphorylation), pyrimidine nucleotide biosynthesis and dTDP sugar biosynthesis. Region V contained modules capable of heme transport, cytochrome c oxidation-reduction (complex IV in oxidative phosphorylation), F-type ATP generation (complex V in oxidative phosphorylation, bacterial type), C5 isoprenoid biosynthesis and lipopolysaccharide biosynthesis. These modules allowed us to further dissect the characteristic metabolic processes exemplified by the KEGG pathways in different samples.
CO2 fixation processes
DBP contained hundreds of reads for the ECs, particularly EC1.2.7.3 (K00174-K00177) and EC1.2.7.1 (K00169-K00172), which are typically involved in autotrophic and anaplerotic CO2 fixation. The two ECs mediate the reaction of succinyl-CoA + CO2 → 2-oxoglutarate (part of the reductive citric acid cycle (rTCA)) and catalyze acetyl-CoA + CO2 → pyruvate (reductive acetyl-CoA reaction; rACA), respectively23. Homologs present in the other samples were not comparable and were ignored. Although there were 55 and 96 reads for K00174 and K00175, respectively, the numbers obtained for CAM were significantly lower than those for DBP (chi-square test; p < 10−4). Occurrence of the two autotrophic reactions in ABP and MED was not likely, as there were <10 reads for these genes.
However, CO2 can be fixed by other steps23. An enzyme encoded by K01595 (EC4.1.1.31) is able to fix CO2 by using phosphoenolpyruvate and another enzyme encoded by K00031 (EC1.1.1.42) consumes 2-oxoglutarate (also part of rTCA). In regions III and IV of Fig. 5, the two functional genes for CO2 fixation were found in CAM and MED. K01595 was enriched in MED as previously reported19, whereas K00031 was more abundant in CAM than in the other samples. Although not as abundant as in MED and CAM, K01595 and K00031 were also identified in DBP and were remarkably under-represented in ABP.
KEGG and SEED annotations of cORFs for ABP and DBP were illustrated by MEGAN21. DBP possessed the Calvin-Benson-Bassham cycle (CBB) for CO2 fixation. There were 19 cORFs for fructose 1.6-bisphosphatase type V (archaeal) and three for type I, whereas only four cORFs were discovered for ABP. Next, we compared some key enzymes in CO2 fixation via the rTCA and rACA pathways. EC1.2.7.3 and EC1.2.7.1 displayed 228 cORFs in DBP, in contrast to six cORFs in ABP. In addition, EC1.1.1.42 and EC4.1.1.31 had 131 cORFs in DBP; a total of 52 such cORFs were identified in ABP. Fixation via these ECs in DBP was supported by abundant cORFs responsible for the supply of acetyl-CoA and phosphoenolpyruvate, which were six to nine-fold more abundant in DBP than in ABP.
Taxonomic assignment of the cORFs for four KEGG genes in DBP is illustrated in Figure 6. These were the most abundant genes in individual steps of autotrophic and anaplerotic CO2 fixation. A total of 66 K00174 cORFs were derived mainly from Archaea and were occupied nearly 50% by the homologs of Thermofilum (Crenarchaeota). Ninety-three K00031 cORFs were evenly distributed across several genera. Two of them, Salinibacter and Meiothermus, belonged to the bacteria and the majority of the remainders were Halobacteriales in the Euryarchaeota. The 38 K01595 cORFs were highly represented by those cORFs from Meiothermus and the 81 K00169 cORFs were largely occupied by Halorhabdus and some methanogens. In ABP, six K00174 cORFs showed a high level of similarity to the homologs from Bradyrhizobium. There were 13 K01595 cORFs, likely belonging to Cupriavidus. The 39 K00031 cORFs were probably associated with Aromatoleum and Cupriavidus.

Taxonomic assignment of CO2 fixation genes in DBP.
The KEGG genes, K00169 (A), K00174 (B), K00031 (C) and K01595 (D) were represented by complete ORFs extracted from reads of DBP. The genera of four functional KEGG genes involved in CO2 fixation were summarized from the best hits obtained in the BLAST results against the NCBI-nr database (E-value <10−4, score >60). Excluding Methanosaeta, the average of the BLAST scores for individual genera exceeded 100.
Anaerobic methane oxidation by Archaea
Because methane dominates the gas mixtures released into ABP and DBP, microbes involved in AOM might play an important role in autotrophy. At present, three AOM archaeal groups (ANME1, ANME2 and ANME3) have been identified: ANME2 and ANME3 belong to the order Methanosarcinales, whereas ANME1 is more distantly related24. The functional gene mcrA (Methyl Coenzyme M Reductase A) was the target in studies of AOM and methanogens. In the metagenomes, a BLAST search facilitated the identification of a few mcrA reads only in DBP. Similarly, clones were successfully obtained from DBP but not from ABP.
Phylogeny of mcrA genes was reconstructed using proteins deduced from the cloned sequences and homologs. Although they were poorly supported by low bootstrap values, 17 McrA cloned protein sequences clustered with two ANME3 McrA proteins identified in the Cascadia margin sediment and in the Arctic sediment25,26 (Fig. 7). Together, they were distant from the other ANME3 McrA proteins. Only one cloned sequence (no. 65) grouped with a known ANME3 McrA protein from Methanohalophilus. These two proteins were much closer to the main branch of ANME3 than those in the 17 cloned sequences. McrA proteins belonging to ANME1 and other cloned sequences were separated from the other clades. The protein sequence from clone no. 43 grouped with the ANME1 McrA sequences; the other 14 McrA sequences from the clones formed an independent internal clade that was distantly related to the ANME1 group. The large dissimilarity of some DBP McrA proteins compared with others confers their unique status among all known mcrA genes and supports their identification as a potentially new ANME group.

Phylogeny of McrA partial sequences.
The rooted maximum likelihood tree was reconstructed using known McrA partial sequences and translated amino acids from mcrA gene cloning sequences. Species names (or sampling site) and amino acid accessions of the sequences are shown. Bootstrap values were based on 1000 replicates; only those with a value > 50% are shown.
Brine and deep-sea water sample-specific COG pathways
With statistical analyses, COG pathways showing a significant difference (p < 0.05) between ABP and DBP were obtained. The DBP metagenome had significantly more genes that participate in biosynthesis pathways, including amino acids, nucleotides, heme, CoA and NAD (Table S2). Active hydrocarbon metabolic processes in DBP were inferred from the presence of significantly enriched pathways involved in glycolysis, gluconeogenesis and carbohydrate transport. The ABP metagenome displayed significant differences in cell wall/membrane/envelope biogenesis, inorganic ion transport and metabolism, cell motility and transcriptional regulators. Other pathways, such as flagellum biogenesis and lipid A biosynthesis, were considered as supplementary to these four large pathways (Table S2).
There were likely pathways involved in the adaptation to deep-sea water or the hydrothermal brine environment. Considering differences in the gene profiles, they were expected to be commonly enriched in both metagenomes from deep-sea water or the brine environment. COG pathways in ABP and DBP were separately compared with those of the deep-sea water metagenomes and commonly enriched pathways in ABP and DBP included the following: menaquinone and ubiquinone biosynthesis, the multi-subunit Na+/H+ antiporter, carbohydrate transport and metabolism and transcription and transcriptional regulators. Conversely, CAM and MED were compared separately with the brine samples and COG pathways that were commonly enriched in the deep-sea water metagenomes comprised the following: F0F1-type ATP synthase subunits, the glyoxylate circle, Na+-transport via NADH:ubiquinone oxidoreductase subunits, the TCA cycle and protein translation, location and modification. The deep-sea samples also featured a variety of biosynthetic activities such as biosynthesis of amino acids, biotin, coenzyme A, heme, terpenoid and the cell wall/membrane/envelope.
Some of these COG pathways are illustrated in Figure 5. However, novel findings were also obtained. The glyoxylate cycle is a shortcut pathway in the TCA cycle and succinate is produced through the use of isocitrate in the shortcut27. Consequently, two CO2 fixation steps in the reductive TCA cycle are bypassed. The strong activity of the glyoxylate cycle in deep-sea water samples was coincident with a low abundance of bypassed genes in the TCA cycle. There was a remarkable enrichment of isocitrate lyase (COG2224) and malate synthase (COG2225) genes in CAM and MED compared to ABP and DBP. Chi-square tests were conducted to compare the two COGs between the brine pool metagenomes and deep-sea water counterparts. Excluding the comparison of COG2224 between ABP and CAM, all of the other comparisons showed a significant difference (chi-square test; p < 10−4). In addition, different types of sodium pumps were detected. In the brine samples, a multi-subunit Na+/H+ antiporter was strongly represented by the corresponding gene, whereas in the deep-sea water samples, Na+-transportation coupled with complex I in oxidative phosphorylation was utilized as a transport pathway28,29. Between the brine metagenomes, both Na+ transport systems were significantly more abundant in DBP (Table S2).
Discussion
In the present study, brine pool metagenomes in the Red Sea were analyzed and compared with deep-sea water metagenomes. The results revealed drastic differences in the composition of the communities and metabolic spectra between the brine pool metagenomes. Although the two metagenomes have been compared in our previous work, only aromatic-degrading bacteria and metabolic pathways were examined to document the presence of aromatic compounds in the Atlantis II brine pool15. The current study elaborates on how the two brine pool ecosystems can be further differentiated via a comprehensive comparison of the microbial communities and their potential functions. In particular, we revealed archaeal taxa and metabolic pathways that have not been previously observed15. The samples from adjacent brine pools revealed that our two brine metagenomes differed even more remarkably than the deep-sea reference metagenomes sampled at widely separate sites and could not be grouped due to their high level of dissimilarity. Considering the historical connection of the two brine pools8, the ecosystem in the Discovery brine pool might have been similar to that in the Atlantis II in the past. The present physical separation likely resulted in decrease in volume of the two brine pools, which has been reported in previous studies9. After the separation of the two brine pools, the exposure of Atlantis II to increasingly strong geothermal activities on the sea floor probably changed the original environment of Atlantis II. For example, in ABP, we did not find methanogens, AOM Archaea or other autotrophic microbes, which were present in DBP. The apparent absence of chemoautotrophic microbes in ABP might have completely converted the mode of life in this pool to heterotrophy. The present study provides evidence that microbes in ABP depended heavily on extracellular organic carbon, including aromatic compounds, non-aromatic heterocyclic compounds, amino acids, storage sugars and structural sugars in the surrounding water. Overall, conditions in the present lower convective layer of Atlantis II brine pool have selected against archaeal autotrophic and methanogenic inhabitants, while imposed minor effects on the bacterial residents. However, the elimination of these autotrophic inhabitants was probably not as extreme as demonstrated in this study. Considering the low cell density in the ABP sample, sampling drift and the experimental approach could have influenced our results. Crenarchaeotic marine group I (1.7%), Methanomicrobia (0.07%), Halobacteria (0.08%) and Archaeoglobi (0.15%) were detected in the lower convective layer of Atlantis II in an independent study (unpublished data). However, diversified archaeal groups were detected in the subsuperficial sediments of ABP by Siam et al. (2012)30; however, their proportion in the whole community was not determined. In the same report, Archaea were also identified in DBP sediment samples, whereas Crenarchaeota dominated the archaeal communities rather than Euryarchaeota, which consisted of the overwhelming majority of Archaea in the overlying brine in the present study. The differences between the two studies may be a consequence of environmental changes in the sediments. Although more Archaea can probably be identified in Atlantis II, it is clear that the archaeal community in that pool has drastically diminished relative to that in the Discovery brine pool, potentially to the edge of extinction.
A considerable fraction of the archaeal community in DBP was believed to be capable of fixing CO2, but these representatives were almost absent from ABP. This difference might be explained by CO2 limitations in ABP. Although in situ CO2 concentration in the Atlantis II brine pool was not measured, a body of evidence from the following geochemical analyses of brine water and sediments has suggested a very low CO2 concentration in Atlantis II. In several sediment cores, manganosiderite minerals were much more abundant in Atlantis II than in all of the other brine pool sediments in the Red Sea1. These minerals were formed by the interaction of Fe2+ and Mn2+ ions with CO2. The sources of CO2 could be the hydrothermal fluids and biogenic CaCO3 that dissolved in the brines at low pH values1. However, in such a scenario, almost all the CO2 would be accepted rapidly by Mn and Fe ions that are injected into the brine pool via geochemical activities. Concentrations of Mn and Fe ions in the lower convective layer of the Atlantis II brine pool were much higher (Fe, 120×; Mn, 28×) than those in Discovery (Ref. 1 and our unpublished data). CO2 in the lower layer of the Atlantis II could be easily fixed by the large amount of Mn and Fe ions and precipitated as manganosiderite in the sediments. Therefore, deprivation of CO2 by metal ions limited CO2-dependent autotrophy, particularly for the archaeal groups as indicated in the present study. In contrast, CO2 fixation might also be conducted by bacteria such as Cupriavidus, Salinibacter and Meiothermus; however, the latter two genera were not found in ABP, perhaps because the extreme environment of the Atlantis II pool inhibited their colonization. This finding might also partially explained by the compositional differences in the bacterial communities between brine metagenomes at the phylum level because Meiothermus and Salinibacter represent Deinococcus-Thermus and Bacteroidetes/Chlorobigroup, respectively. Hence, the CO2 limitation in Atlantis II likely also accounted for the elimination of CO2 autotrophic Archaea and some bacterial inhabitants. Regarding Cupriavidus spp. in ABP, the question is whether the involved genes were present to fix CO2 or to function in the reverse direction. These genes play a role in several different metabolic pathways. It is also a possibility that the genes were not expressed under low CO2 concentrations. Furthermore, Cupriavidus can degrade aromatic compounds31 and thus, their life mode might be heterotrophic in Atlantis II where organic maturation continues to occur15. The major role of Cupriavidus in Atlantis II was probably metal precipitation, as it is well known that Cupriavidus spp. are capable of precipitating different metal ions32,33.
CO2 fixation was also reported in a recent metagenomic study examining a Mediterranean deep-sea brine lake (DHAL)34. DBP and DHAL brine samples shared some taxonomic microbial assemblages such as Firmicutes and Gammaproteobacteria. Moreover, almost all types (CBB, cTCA and cACA) of CO2 fixation uncovered in this study were performed by Euryarchaeota and Gammaproteobacteria present in the DHAL brine sample (a phosphoenolpyruvate-related type of CO2 fixation was not described)34. However, the presence of CO2 autotrophic Crenarchaeota in DBP is an important difference between these two deep-sea brine pools.
High concentrations of methane derived from hydrothermal fluids have been reported for the Atlantis II pool13,35. In such an environment, methane is normally consumed by AOM from the ANME3 group, which uses manganese oxide as an oxygen donor36. Because Discovery pool may be less disturbed by hydrothermal fluids, manganese oxides could be maintained at the sub-floor and likely be supplied by those manganese oxides generated in Atlantis II that spread into Discovery via deep-sea water circulation around the Chain brine pool (refer to the deep-sea water channel in Fig. 1). As a result, the lower convective layer of Discovery could support the rapid assimilation of methane by ANME3, thus explaining the presence of ANME3 methanotrophic Archaea in DBP. The case differs in Atlantis II, where manganese ions are still oxidized at the interface between the deep-sea water and the brine pool1. Manganese oxides then transfer the oxygen to ferrous ions in the upper convective layer, especially at the boundary between the lower and upper brine layers of the brine pool1, resulting in the large-scale precipitation of ferrous oxides rather than manganese oxides to the sub-floor. There is evidence that Mn-hydroxides do not accumulate at the bottom of this pool as they do in Discovery, but rather, they have fallen to the slopes around the interface along the periphery of the Atlantis II brine pool and even in Discovery1. Hence, methane oxidation by ANME3 might occur at the interface of the Atlantis II pool and the seawater. As expected, mcrA sequences identical to those of ANME3 in DBP had been successfully cloned using the water sample collected from the interface of Atlantis II (unpublished data). Although Fe hydroxide also can be potentially used for methane oxidation by ANME3, Mn hydroxide is preferred due to its five-fold higher potential for free energy gain36. Therefore, the interface between the Atlantis II brine pool and the deep-sea water was found to be a better niche for ANME3. In addition, a novel branch was identified in our phylogenetic tree. Because no mcrA cloning sequences clustered with the known methanogens, they were likely derived from methanogenic Archaea. Without additional evidence, however, their role in ABP remains unclear.
However, methane can be consumed by methanotrophic microbes that possess monooxygenase (encoded by the pmo and mmo genes)37. We used monooxygenase proteins to search for possible homologs in the four metagenomes. Positive results (BLAST cutoff score of 100) were obtained only for the CAM metagenome, in which uncultured crenarchaeota and methanotrophic bacteria (Methylococcaceae) were candidates for the consumption of methane under aerobic conditions. A high expression level of pmo genes was confirmed in a recent metatranscriptomic study of a seawater sample from the Carmen Basin, Gulf of California20.
The presence of aromatic compounds in the lower convective layer of Atlantis II has been reported in our previous work15. In theory, the increasing temperature in the Atlantis II brine pool has converted organic carbons into aromatic compounds15. The change in carbon sources under high temperatures can affect microbial communities and metabolic activities dramatically. Aromatic compounds have seldom been reported as carbon sources for Archaea. For methanogens, alkanes, acetate and volatile fatty acids are typically used to produce methane38,39,40. If the methanogens were the original inhabitants of Atlantis II, sufficient labile carbon was not easy to access in the lower convective layer. Therefore, the exclusion of archaeal methanogens is likely ascribed to the conversion of organic carbon into aromatic compounds. This would also have limited the diversity of the bacterial communities because only those capable of degrading aromatic compounds could proliferate in the lower convective layer of Atlantis II.
We have argued that carbon sources contributed to the compositional shift in ABP. Environmental stresses imposed by chemocline, thermocline, halocline and heavy metal content could have greatly impacted the microbes. To cope with chemocline and thermocline, microbes may employ chemotactic mechanisms to sense surrounding environmental changes; Bacteria and Archaea can react to changes in chemical factors by switching their motility, speed and the direction of their flagella41. ABP possessed a greater number of chemotactic genes; for example, it had four times more mcp (methyl-accepting chemotaxis protein) genes compared with DBP. More detailed comparisons between ABP and DBP with respect to chemotactic genes and pathways have been reported in our previous work15. Surprisingly, in the current study, related pathways for flagellar assembly and bacterial chemotaxis demonstrated greater enrichment in MED than in all of the other metagenomes evaluated. The significant enrichment for the chemotaxis in MED was considered to be an adaptation to the extreme lack of nutrients in Mediterranean deep-sea water (Ref. 19 and references therein). One may argue that the pursuit of nutrients is even more important than the stresses imposed by chemocline and thermocline in the lower convective layer of the Atlantis II pool. Perhaps, the enrichment of chemotactic genes in ABP relative to DBP was also due to a shortage in the nutrient supply.
Two approaches, via the NADH:ubiquinone Na+ antiporter and the multi-subunit Na+/H+ antiporter, were discovered in the four metagenomes. First, multi-subunit Na+/H+ antiporter genes were significantly enriched in both of the brine metagenomes. Because the salinity of the sampling sites in the brine pools was much higher than that in the deep-sea waters (34.6‰ for CAM and 38.7‰ for MED), the multi-subunit Na+/H+ antiporter approach likely played a more important role in microbial adaptation to extreme salinity. Second, the comparison between the two brine metagenomes revealed that both antiporters were more strongly represented by the reads in DBP. Despite the same salinity at the two sites, this result suggested that microbes in DBP, surprisingly, possessed more Na+ transporter genes than did those in ABP. The NADH:ubiquinone Na+ antiporter is the dedicated Na+ pump in many aerobic bacteria42. As expected, this pump was significantly enriched in the deep-sea water metagenomes in the present study, as compared with ABP and DBP. The pumping process is supposed to be coupled to the strong oxidative phosphorylation that occurs in deep-sea water metagenomes and, therefore, to serve as an adaptive strategy in oxic seawater environments. Furthermore, a large number of ATPs generated by the oxidative phosphorylation process could drive various biosynthetic activities, thus explaining the enrichment of biosynthesis genes in deep-sea water metagenomes.
Considering the ongoing strong metal precipitation activities1, the Atlantis II brine pool imposes a stronger impact on microbial inhabitants that are exposed to a large amount of metal ions and a higher temperature, compared with those present in the Discovery. However, in DBP, we identified more metal ion pumps for cobalt, nickel, manganese and iron, which suggests that these metal transport processes are important for the microbes in DBP. In addition, there was a lack of a correspondingly high abundance of metal transporter genes in ABP, although manganese and iron were the major types of metal ions present in the lower convective layer of Atlantis II1. The microbes in ABP elected energy-saving mechanisms, likely because these pumps are energy-dependent ABC transporters. This phenomenon was exemplified by a two-component copper transport system encoded by the cus operon that had significantly more genes in ABP than in DBP.
Most of the adaptive mechanisms discussed above may explain the adaption observed for the microbes in DBP. However, the mechanism by which microbes adapted to the complex and extreme conditions in the Atlantis II brine pool remains unclear. To address this issue, the enriched pathways in ABP were further examined and their possible adaptive roles were explored. Transcriptional regulators were clearly important for a rapid response to the fluctuation of variant factors in the Atlantis II brine pool. The enrichment of genes for secretion systems IV and VI in ABP also suggested a potentially important mechanism. Six types of secretion systems have been identified thus far43. The secretion system IV has been recently reported in thermophilic bacteria and is thought to govern the transfer of DNA and protein between cells44.
Because our conclusions are based on analyses of metagenomes from a small volume of brine water, further transcriptomic studies are required to support our findings regarding autotrophic metabolism. A comprehensive survey of microbial communities should be undertaken at more sampling sites with replicates to determine whether the microbial communities changed dramatically in the different layers and at different sites in the same layers of the brine pools.
Methods
Sampling
Water samples were collected from the Atlantis II and Discovery brine pools during an R/V OCEANUS cruise in October 200816. Both samples were obtained from the lower convective layers of the pools (>2100 m). At each sampling site, 4 L of brine water was immediately filtered through a 1.6 μm GF/A membrane (diameter 125 mm; Whatman, Clifton, NJ) followed by a 0.22 μm Steripak-GP polyethersulfone membrane (Millipore, Bedford, MA) to capture prokaryotic cells. Total genomic DNA was extracted and purified according to the SDS-based method described by Huber et al. (2002)45. Temperature, depth and salinity were recorded by a CTD unit.
Microbial cell counting
Three replicates of each 10-mL sample were immediately fixed with formaldehyde. The fixed samples were then filtered through polycarbonate membranes (0.2 μm, 25 mm diam., Isopore; Millipore, Bedford, MA) and stained with 4,6-diamidino-2-phenylindole (DAPI) for 15 min in the dark. Cells were counted in 10 randomly selected fields using an Olympus epifluorescence microscope (FSX100, Hamburg, Germany).
Metagenome analyses
Pyrosequencing of the two brine pool metagenomes was performed as described previously15. Because no deep-sea brine metagenomes were available in the public databases, two reference deep-sea water metagenomes were obtained from the CAMERA (camera.calit2.net) and NCBI-SRA (www.ncbi.nlm.nih.gov/sra) databases. CAM (accession ID: CAM_S_465 in CAMERA) was sampled near a deep-sea hydrothermal vent in Carmen Basin, Gulf of California (26°38′N, 110°72.8′W). The depth, temperature and salinity of the sampling site were 1900 m, 2.6°C and 34.6‰, respectively. MED is referred to as SRX087122 in the NCBI-SRA. MED was sampled at 4,908 m in the Matapan-Vavylov Deep, Mediterranean Sea at (36°34.00'N, 21°07.44'E)19. The temperature and salinity were 14.3°C and 38.7‰, respectively. Because the microbial community in MED was relatively simple19, only a small portion of the MED metagenome (557,800 reads) was used in the present study. The reads were randomly selected from the original file.
16S rRNA gene fragments in the reads were identified using Meta-RNA46 and extracted with an in-house program. Using cdhit-45447, duplications that occupied 20–30% of the fragments were removed and the longest representatives among the duplicates were retained. Taxonomic classification of the fragments was conducted with the RDP naïve Bayesian Classifier version 2.5 (http://rdp.cme.msu.edu/). A confidence threshold of 50% was applied to the classification at phylum and genus levels48. Because Ralstonia and Wautersia have been jointly combined with Cupriavidus49, the percentage of Cupriavidus was the total percentage of the three genera. The protocol used herein contained several modifications compared with that introduced in our previous work15, but the percentage of dominant bacterial genera such as Cupriavidus remained almost the same.
Pyrosequencing reads were used to BLAST against the two protein databases, KEGG (version 51)50 and COG (http://string-db.org), using an e-value of <10−4. The results of the BLAST search were summarized by selecting the best hits as KEGG genes or COGs in the reads. EC numbers were also extracted from the KEGG gene annotation. Redundant reads from pyrosequencing emPCR amplification and whole genome amplification were filtered using an in-house tool. The algorithm employed for the redundancy clearance was as follows: reads for the same KEGG genes and COGs were aligned pairwise and evaluated for identical portions (in a size of 50 bp) on both strands. The identical portions did not occur exactly at the first position of the two reads and could be shifted slightly. Shorter reads were removed if they contained portions identical to those in one of the longer reads.
The numbers of KEGG genes, COGs and ECs were standardized separately. A Bray-Curtis dissimilarity matrix was calculated from each of the three datasets. AHC analysis was performed using the pvclust package in the R project. Ward's method was employed for agglomerative hierarchical clustering. In addition, the dissimilarity matrix was used in the PCoA analysis.
The KEGG dataset was then normalized using a z-score and the genes were clustered using Cluster351. A complete linkage method and a metric of correlation (uncentered) were employed during the clustering of samples and genes. The KEGG genes in the five sample-specific gene clusters shown in Figure 5 were sorted into different KEGG pathways. In Figure 5, only pathways with more than 10 genes or those with at least 50% of all the genes in a given pathway are displayed. However, some pathways contained genes that were shared with some of the other pathways. To avoid over counting such genes, at least two pathway-specific genes were identified in a selected pathway. The positions of the genes in the KEGG pathway maps were rechecked to confirm their involvement in the essential function of a given pathway. Sorting of the genes into KEGG functional modules was performed using the same approach. In the modules selected for comparison, at least 50% of their KEGG genes had been identified in the metagenomes.
We did not conduct homopolymer error correction for the four metagenomes. For the brine pool metagenomes, the cORFs were extracted from reads longer than 200 bp and defined as those covering whole reads without interruptions except for the last five positions. This strategy may preclude the use of reads with a premature termination of the ORF, which often occurs in pseudogenes or reads with a low pyrosequencing quality. A BLAST search was performed against the NCBI protein database and the taxonomies for the resultant hits (score >60) were summarized using MEGAN21. The KEGG and SEED genes assigned to different pathways could also be visualized through the use of MEGAN. Using ShotgunFunctionalizeR in the R project, the COGs in the cORFs were compared to identify genes and pathways that differed significantly in relative abundance. P-values were adjusted using the Bonferroni correction.
Phylogeny reconstruction using cloned mcrA genes from DBP
Forward and reverse primers used for PCR amplification of mcrA genes were 5′-TAYGAYCARATHTGGYT-3′ and 5′-ACRTTCATNGCRTARTT-3′, respectively52. Each cycle (total of 30 cycles) consisted of a 60 s denaturation step at 94°C, a 45 s annealing step at 45°C and a 50 s elongation step at 72°C. Each PCR reaction (total of 25 μl) contained 2.5 μL of 10XPCR buffer, 2 μL of 2 mM dNTP and 1 U of Taq DNA Polymerase (Takara, Tokyo, JP). The PCR products were purified and cloned using the TOPO-TA Cloning Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. The sequences were deposited in NCBI (accession numbers: JX907770-JX907800). Next, the translated proteins were aligned with other McrA protein sequences collected from NCBI using MUSCLE3.553 . The alignment was manually checked to remove gaps and was then used to reconstruct a neighbor-joining (NJ) tree with 1000 bootstrap replicates using MEGA554. A maximum likelihood (ML) phylogenetic tree with 1000 bootstrap replicates was then built, using the NJ tree as an internal tree. Jones_Taylor_Thornton was selected as the substitution model. McrA sequences from methanogens served as the outgroup for the rooted ML tree.
References
Gurvich, E. G. In: Metalliferous Sediments of the World Ocean (Springer Berlin Heidelberg, 2006).
Ramboz, C., Oudin, E. & Thisse, Y. Geyser-type discharge in Atlantis II Deep, Red Sea: evidence of boiling from fluid inclusions in epigenetic anhydrite. Can Mineral 26, 765–786 (1988).
Oudin, E., Thisse, Y. & Ramboz, C. Fluid inclusion and mineralogical evidence for high temperature saline hydrothermal circulation in the Red Sea metalliferous sediments: preliminary results. Mar. Mining 5, 3–31 (1984).
Miller, A. R. et al. Hot brines and recent iron deposits in deeps of the Red Sea. Geochim Cosmochim Acta 30, 341–350 (1966).
Girdler, R. W. A review of Red Sea heat flow. Phil Trans Roy Soc Lon A 267, 191–203 (1970).
Schoell, M. & Hartmann, M. Detailed temperature structure of the hot brines in the Atlantis II Deep area (Red Sea). Marine Geol 14, 1–14 (1973).
Blanc, G. & Anschutz, P. New stratification in the hydrothermal brine system of the Atlantis II Deep, Red Sea. Geology 23, 543–546 (1995).
Turner, J. S. A physical interpretation of the observations of hot brine layers in the Red Sea. (Springer, Berlin; 1969).
Monnin, C. & Ramboz, C. The anhydrite saturation index of the ponded brines and sediment pore waters of the Red Sea deeps. Chem Geol 127, 141–159 (1996).
Ross, D. A. & Hunt, J. M. Third brine pool in the red sea. Nature 213, 687–688 (1967).
Swallow, J. C. & Crease, J. Hot salty water at the bottom of the Red Sea. Nature 205, 165–166 (1965).
Hartmann, M., Scholten, J. C., Stoffers, P. & Wehner, F. Hydrographic structure of brine-filled deeps in the Red Sea--new results from the Shaban, Kebrit, Atlantis II and Discovery Deep. Marine Geology 144, 311–330 (1998).
Faber, E. et al. Methane in Red Sea brines. Organic Geochem 29, 363–379 (1998).
Anschutz, P. & Blanc, G. Heat and salt fluxes in the Atlantis II Deep (Red Sea). Earth Planet Sci Lett 142, 147–159 (1996).
Wang, Y. et al. Hydrothermally generated aromatic compounds are consumed by bacteria colonizing in Atlantis II Deep of the Red Sea. ISME J 5, 1652–1659 (2011).
Swift, S. A., Bower, A. S. & Schmitt, R. W. Vertical, horizontal and temporal changes in temperature in the Atlantis II and Discovery hot brine pools, Red Sea. Deep Sea Res I 64, 118–128 (2012).
Cole, J. R. et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucl. Acids Res. 37, 141–145 (2009).
Dick, G. J. & Tebo, B. M. Microbial diversity and biogeochemistry of the Guaymas Basin deep-sea hydrothermal plume. Environ Microbiol 12, 1334–1347 (2010).
Smedile, F. et al. Metagenomic analysis of hadopelagic microbial assemblages thriving at the deepest part of Mediterranean Sea, Matapan-Vavilov Deep. Environ Microbiol 15, 167–182 (2012).
Lesniewski, R. A., Jain, S., Anantharaman, K., Schloss, P. D. & Dick, G. J. The metatranscriptome of a deep-sea hydrothermal plume is dominated by water column methanotrophs and lithotrophs. ISME J 6, 2257–2268 (2012).
Huson, D. H., Mitra, S., Ruscheweyh, H.-J., Weber, N. & Schuster, S. C. Integrative analysis of environmental sequences using MEGAN4. Genome Res 21, 1552–1560 (2011).
Pitcher, R. S. & Watmough, N. J. The bacterial cytochrome cbb3 oxidases. Biochim. Biophys. Acta 1655, 388–399 (2004).
Hügler, M., Huber, H., Stetter, K. O. & Fuchs, G. Autotrophic CO2 fixation pathways in archaea (Crenarchaeota). Arch Microbiol. 179, 160–173 (2003).
Knittel, K. & Boetius, A. Anaerobic oxidation of methane: progress with an unknown process. Ann Rev Microbiol 63, 311–334 (2009).
Hallam, S. J., Girguis, P. R., Preston, C. M., Richardson, P. M. & DeLong, E. F. Identification of methyl coenzyme M reductase A (mcrA) genes associated with methane-oxidizing archaea. Appl Environ Microbiol 69, 5483–5491 (2003).
Losekann, T. et al. Diversity and abundance of aerobic and anaerobic methane oxidizers at the Haakon Mosby mud volcano, Barents Sea. Appl Environ Microbiol 73, 3348–3362 (2007).
Kornberg, H. L. The role and control of the glyoxylate cycle in Escherichia coli. Biochem J. 99, 1–11 (1966).
West, I. C. & Mitchell, P. Proton/sodium ion transport in Escherichia coli. Biochem. J 144, 87–90 (1974).
Stolpe, S. & Friedrich, T. The Escherichia coli NADH:Ubiquinone oxidoreductase (complex I) is a primary proton pump but may be capable of secondary sodium antiport. J Biol Chem 279, 18377–18383 (2004).
Siam, R. et al. Unique prokaryotic consortia in geochemically distinct sediments from Red Sea Atlantis II and Discovery Deep brine pools. PLoS ONE 7, e42872 (2012).
Pérez-Pantoja, D., De la Iglesia, R., Pieper, D. H. & González, B. Metabolic reconstruction of aromatic compounds degradation from the genome of the amazing pollutant-degrading bacterium Cupriavidus necator JMP134. FEMS Microbiol Rev. 32, 736–794 (2008).
Lykidis, A. et al. The complete multipartite genome sequence of Cupriavidus necator JMP134, a versatile pollutant degrader. PLoS ONE 5, e9729 (2010).
Reith, F. et al. Mechanisms of gold biomineralization in the bacterium Cupriavidus metallidurans. Proc Natl Acad Sci USA 106, 17757–17762 (2009).
Ferrer, M. et al. Unveiling microbial life in the new deep-sea hypersaline Lake Thetis. Part II: a metagenomic study. Environ Microbiol 14, 268–281 (2011).
Schmidt, M. et al. High-resolution methane profiles across anoxic brine-seawater boundaries in the Atlantis-II, Discovery and Kebrit Deeps (Red Sea). Chem Geol 200, 359–375 (2003).
Beal, E. J., House, C. H. & Orphan, V. J. Manganese- and iron-dependent marine methane oxidation. Science 325, 184–187 (2009).
Lipscomb, J. D. Biochemistry of the soluble methane monooxygenase. Ann Rev Microbiol 48, 371–399 (1994).
Zengler, K., Richnow, H. H., Rossello-Mora, R., Michaelis, W. & Widdel, F. Methane formation from long-chain alkanes by anaerobic microorganisms. Nature 401, 266–269 (1999).
Sorensen, J., Christensen, D. & Jorgensen, B. B. Volatile fatty acids and hydrogen as substrates for sulfate-reducing bacteria in anaerobic marine sediment. Appl. Environ. Microbiol. 42, 5–11 (1981).
Zinder, S. H. In: Methanogenesis: ecology, physiology, biochemistry and genetics. (ed. J. G. Ferry ) (Chapman and Hall, New York; 1993).
Lux, R. & Shi, W. Chemotaxis-guided movements in bacteria. Critical Rev Oral Biol Med 15, 207–220 (2004).
Hase, C. C., Fedorova, N. D., Galperin, M. Y. & Dibrov, P. A. Sodium ion cycle in bacterial pathogens: evidence from cross-genome comparisons. Microbiol Mol Biol Rev 65, 353–370 (2001).
Economou, A. et al. Secretion by numbers: protein traffic in prokaryotes. Mol Microbiol 62, 308–319 (2006).
César, C. E. et al. Unconventional lateral gene transferin extreme thermophilic bacteria. Int Microbiol 14, 187–199 (2011).
Huber, J. A., Butterfield, D. A. & Baross, J. A. Temporal changes in archaeal diversity and chemistry in a mid-ocean ridge subseafloor habitat. Appl. Environ. Microbiol. 68, 1585–1594 (2002).
Huang, Y., Gilna, P. & Li, W. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25, 1338–1340 (2009).
Niu, B., Fu, L., Sun, S. & Li, W. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinformatics 11, 187 (2010).
Claesson, M. J. et al. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 4, e6669 (2009).
Vandamme, P. & Coenye, T. Taxonomy of the genus Cupriavidus: a tale of lost and found. Int J Syst Evol Microbiol 54, 2285–2289 (2004).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucl Acids Res 28, 27–30 (2000).
Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95, 14863–14868 (1998).
Kendall, M. M. et al. Diversity of archaea in marine sediments from Skan Bay, Alaska, including cultivated methanogens and description of Methanogenium boonei sp. nov. Appl Environ Microbiol 73, 407–414 (2007).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32, 1792–1797 (2004).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol Biol Evol 28, 2731–2739 (2011).
Acknowledgements
The authors are grateful to Prof. Paul Harrison for his comments on the manuscript. This study was supported by the National Basic Research Program of China (973 Program, No. 2012CB417304) and an award (SA-C0040/UK-C0016) granted to P.Y. Qian from the King Abdullah University of Science and Technology.
Author information
Authors and Affiliations
Contributions
Y.W., O.O.L. and P.Y.Q. wrote the main manuscript. H.L.C., A.A.S. and G.S.Z. performed the experiments. Y.W. and H.L.C. analyzed the data. Y.W., O.O.L., S.B., A.A.S. and P.Y.Q. designed and prepared the samples. All authors discussed and reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
suppl.tables and figure s1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Wang, Y., Cao, H., Zhang, G. et al. Autotrophic Microbe Metagenomes and Metabolic Pathways Differentiate Adjacent Red Sea Brine Pools. Sci Rep 3, 1748 (2013). https://doi.org/10.1038/srep01748
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01748
This article is cited by
-
Metabolic traits of an uncultured archaeal lineage -MSBL1- from brine pools of the Red Sea
Scientific Reports (2016)
-
Viruses-to-mobile genetic elements skew in the deep Atlantis II brine pool sediments
Scientific Reports (2016)
-
Global diversity and biogeography of deep-sea pelagic prokaryotes
The ISME Journal (2016)
-
Production of halophilic proteins using Haloferax volcanii H1895 in a stirred-tank bioreactor
Applied Microbiology and Biotechnology (2016)
-
Species sorting during biofilm assembly by artificial substrates deployed in a cold seep system
Scientific Reports (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
