
Abstract
Host genotype and gender are among the factors that influence the composition of gut microbiota. We studied the population structure of gut microbiota in two lines of chickens maintained under the same husbandry and dietary regimes. The lines, which originated from a common founder population, had undergone 54 generations of selection for high (HW) or low (LW) 56-day body weight and now differ by more than 10-fold in body weight at selection age. Of 190 microbiome species, 68 were affected by genotype (line), gender and genotype by gender interactions. Fifteen of the 68 species belong to Lactobacillus. Species affected by genotype, gender and the genotype by gender interaction, were 29, 48 and 12, respectively. Species affected by gender were 30 and 17 in the HW and LW lines, respectively. Thus, under a common diet and husbandry host quantitative genotype and gender influenced gut microbiota composite.
Similar content being viewed by others


Introduction
The gut microbiota is a complex ecosystem that has a symbiotic relationship with its host. Their interactions affect the physiological, immunological and nutritional status of the host. A key question in understanding interactions between the host and gut microbiota is if the genetic background and gender of the former influences the population structure of gut microbiota when fed a common diet. The role of host genetics on shaping this vital ‘microbial organ’ is not clear1. Although environmental2 and maternal effects3,4,5 can affect the composition of the microbiota, results from human twin studies are equivocal6,7. Evidence from zebra fish and mice showed that the host influences the diversity and population of gut microbiota8. Moreover a single gene difference in the host can affect the population structure of gut microbiota4,9,10,11. The genotype of the host may affect its microbiota composition either directly through secretions into the gut, control of gut motility and modification of epithelial cell surfaces, or indirectly, through food and lifestyle preferences. These effects are likely to be small and detecting them will require well controlled effects other than those of the host genotype. Thus, choosing a model organism maintained in essentially an identical environment with less maternal effects could enhance our understanding host genotype effects on gut microbiota.
Most host phenotypes involve complex traits that are controlled by multiple genes involving gene networks. There is a dearth of research related to quantitative genetic influences on gut microbiota. In order to address this issue and reduce noise, we conducted an experiment using two lines of chickens that had undergone long term divergent selection for the single trait, 56-day high (HW) or low (LW) body weight (Fig. 1). The lines originated from a common founder population and have complete pedigrees12,13. Throughout all generations, they have been maintained at the same location and reared on the same diets. Selection has resulted in more than a 10-fold difference between them for body weight at selection age. The maximum inbreeding coefficients (F) are 0.53 and 0.61, with a mean of 0.26 (SD 0.15) and 0.30 (SD 0.17) in LW and HW lines, respectively13. QTL mapping revealed 13 loci affecting growth in these two lines, however each locus explained only a small additive effect for this large phenotypic difference14. This moderate F combined with QTL results suggest that there is genetic diversity within each line and the 10-fold body weight difference is because of accumulation of quantitative genes instead of single gene mutation. The detailed information of these two lines was provided in the supplemental section of description of high and low body weight lines of chickens. Aided by next generation sequencing technology, we investigated the population structure of the gut microbiota in adults of these two lines to address if the quantitative genetic background of the host influences gut microbiota.

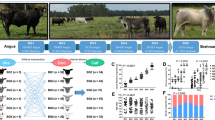
Generation means at selection age (56-day body weight) for high (HW) and low (LW) line females and the distribution for the gut microbiomes composite for HW and LW lines at the phylum level.
Results
Fecal samples from 56 chickens which were comprised of 12 LW females, 14 LW males, 15 HW females and 15 HW males were collected. In order to compare which 16S rRNA variable regions are more suitable to be used for identify taxonomy from chicken feces, a pilot experiment was performed. DNA from 5 fecal samples from each group of LW females, HW males and HW females, totally 15 samples were used in the pilot experiment. Fragments encompassing V3, V4, V1–V3 and V4–V6 16S rRNA hypervariable regions were PCR amplified from each of those 15 DNA samples. PCR production of V3 and V4 were sequenced by Illumina miseq and V1–V3 and V4–V6 were sequenced by Roche's GS FLX+. Taxonomy at species rank was classified by best hit classification function in MGRAST15. We identified 57, 190, 159, 97 species in at least three samples by V3, V4, V1–V3, V4–V6, respectively. These results are consistent with those showing that V3 underestimates species richness and V4 provides estimates similar to those obtained with the nearly full-length fragment16. The number of species identified by combination of these hypervariable regions are presented in Supplementary table S5. Thus, V4 of 16S rRNA were used for the rest of our study. A total of 1,141,781 of V4 16S rRNA sequences reads from the 56 samples with an average of 20,389 sequences reads for each sample (the minimum # of one sample was 7,590 and the maximum was 32,877) were used for this project. The average length of sequence reads was 228 bp and they were classified into different taxonomy using MGRAST15 (http://metagenomics.anl.gov/). The taxon abundance of each sample was generated into phylum, class, order, family, genera and species levels using mainly database of RDP, aided by Greengene and SSU databases. Taxonomies present in at least 14 samples were considered as common and their abundance count of samples were used for further analysis. At the phylum level, the % distribution of microbiome community of HW and LW chickens, respectively was 38 and 37 for Firmicutes, 31 and 33 for Proteobacteria, 18 and 18 for Actinobacteria, 11 and 10 for Bacteroidetes and less than 1 for Deinococcus-Thermus, Euryarchaeota and Fusobacteria (Fig. 1). At the phylum level, the lines were similar (p > 0.05) for fecal microbiome microbial community structure; a pattern that was consistent until the family level (Fig. 1 and Supplementary Fig. S1). Because both lines originated from the same founder population with the major difference being that they were selected 54 generation for high or low body weight at 56 days of age, a trait controlled by quantitative genes, suggests the limitation of host quantitative genetic background influences on the population structure of gut microbiome from the phylum to family levels.
Diversity of the fecal microbiome microbial community at the genera and species level
To further characterize the changes in microbiota imposed by the host genotype, 16S rRNA were classified taxonomically to the genera and species levels. For statistical analysis to detect effects by host quantitative genotype and gender, 103 genera were used. Of these 103 genera, 38 were affected by genotype, gender and genotype by gender interactions (p < 0.05). Among these genera, 16 genera were affected by genotype, 26 by gender and 8 by the genotype by gender interaction (Supplementary Table S3). Principle component analysis (PCA) of HW and LW samples of the 16 genera did not clearly separate the lines. Therefore, for further analysis, 190 species were used and 68 were affected by genotype, gender, or genotype by gender interactions (p < 0.05). Species affected by genotype (p < 0.05), gender and genotype by gender interactions were 29, 48 and 12, respectively (Table 1). Using the 29 significantly affected species, it was possible to separate a majority of the LW from the HW samples (Fig. 2b) demonstrating a host quantitative genotype influence on the microbiome population structure.

Gut microbiota compositions for two lines (HW and LW) of chickens with different quantitative genotypes.
(a) Distribution of gut microbiomes composite for HW females, HW males, LW females and LW males at the genera level. (b) The principal components analysis (PCA) plot of 56 samples from HW and LW lines using 29 gut microbiome species which were affected by quantitative genetics background. (c) PCA plot of 56 samples from HW and LW lines using 11 gut microbiome species belong to Lactobacillus which were affected by gender. (d) PCA plot of 29 samples from HW and LW males using 8 gut microbiome species which were affected by quantitative genetic background. (e) PCA plot of 27 samples from HW and LW females using 20 gut microbiome species which were affected by quantitative genetic background.
Because there are significant physiological and biological functions between adults, within line comparisons were made between genders. There were 20 and 8 species that differed for HW and LW females and males, respectively (Table 2) and the samples could be separated into categories which matched their host quantitative genotype (Fig. 2d, e). The separation was clearer for females than males suggesting that while the host quantitative genotype influenced the population structure of gut microbiota, within a line the target species were not necessarily the same for females and males.
Gender has a significant effect on gut microbiota composition
Forty-eight species were affected (p < 0.05) by gender with the pattern being line dependent (Table 1 and Supplementary Fig. S2). That is, 30 species were different (p < 0.05) between males and females in the HW line (Supplementary Table S1 and Supplementary Fig. S2) and 17 species in the LW line according to T-test (Supplementary Table S2 and Supplementary Fig. S2). Our data are from adult chickens where the females have a large demand for egg production. This major biological difference between adult males and females could easily contribute to gender differences in gut microbiota composition. The large difference between lines for body weight and their maintenances reflect a situation in which the daily food intake is much less for LW than HW individuals. However, their mean percent egg production is similar (59 ± 11 in HW and 48 ± 13 in LW) (Supplementary Table S4) which could explain the line by gender interaction.
Lactobacillus is a major diversity genera
Of the 68 significantly different abundance of species, 15 belong to Lactobacillus. From these 15 species via PCA we could identify LW females from all samples (Fig. 2c). Of the 20 species that were different (p < 0.05) between HW and LW females (Table 2), 6 belong to Lactobacillus. Tissue growth and reproduction are energy-dependent processes and Lactobacillus is involved in metabolites of bile acids which are related obesity and the metabolic syndrome17,18,19,20,21,22, which also plays a key role in metabolites of phenolic, benzoyl and phenyl derivatives which are related to weight loss23,24. Moreover, Lactobacillus could affect lipid metabolites which impact intestinal permeability and activate the intestine-brain-liver neural axis to regulate glucose homeostasis25,26. Thus Lactobacillus may impact LW females in food digestion, energy homeostasis and energy-related metabolites in order to achieve the similar egg production even though their food intake is relatively less on a body weight basis than that for the HW line.
Host genetic factors shape individual microbiome species diversity
We agree that abundance of each species in gut microbiota can be treated as quantitative trait which is influenced by both environmental and host genetic factors5. In an effort to distinguish the proportion of host genetics factors in shaping individual microbiome species diversity and because the chickens used in this experiment were pedigreed, we calculated the heritability for 23 species (13 belong to Lactobacillus) using REML algorithm by wombat27 (Table 1). Although some of the heritabilities were high, none was significantly different from 0. This may be because the size of population was not large.
Discussion
In individual vertebrates, the species composition and their relative proportion in gut microbial communities vary and are influenced by both the environmental and genetic background of the host. Because external factors such as diet, husbandry, maternal and litter effects can influence this complex ecosystem, they could dilute or mask the impact of the host's genetic background. Thus, in order to study the host quantitative genetic background that may influence the composition of the gut microbiota, we needed to control for these environmental factors. In this paper, the HW and LW lines of chickens are used as a model. These lines originated from a common founder population and were maintained in a similar environment for the 50+ generations with the variable being selection for either high or low 56-day body weight. Thus, they address the impact of host quantitative genetic background to the population structure of the gut microbial community.
We treated abundance of species in the gut microbial community as a quantitative trait to address how host background influenced the composition of gut microbiota. Using this strategy, it is obvious that variation in some species is influenced by the quantitative genetic background of the host. The pattern of host genetic influence is different in adult males and females demonstrating gender as a factor that impacts the composition of gut microbiota. Of host-microbe interactions, Lactobacillus is a major genera suggesting that Lactobacillus plays a key role related to phenotype of body weight28. Lactobacillus has a long history as an exogenous probiotic. In our study, we also found that LW females had differing levels of Lactobacillus compared to HW females. Recently, it was reported that feeding laying hens with 0.6% metabolite combinations of Lactobacillus in the diet improved egg production29. Also, some species of Lactobacillus accelerated gonadal differentiation in zebrafish30. Although lacking of direct evidence showing how specific species in Lactobacillus contribute to egg production in LW, it is possible that there is an association.
Methods
Animals and sample collection
Protocols used for this experiment were consistent with those approved by the Institutional Animal Care and Use Committee at Virginia Tech. At 245 days of age fecal samples were obtained randomly from 15 males and 15 females from generation 54 of the HW and LW lines. These 60 individuals were random samples from the larger population of their generation which had been under similar husbandry conditions. All were housed in individual cages (dimensions of 48 × 28 × 46 cm) in the same room with at 14:10 light:dark photoperiod. By keeping each chicken in an individual cage with wire floors, we prevented uncontrolled particle intake as well as feathers which may influence microbiota31. Chickens were fed a corn-soybean non-pathogen free diet in mash form. Details are provided in supplement of breeder diet ingredients. Cages had sloping wire floors with papers beneath them to collect feces. At 10:00 am papers beneath the cages were replaced and during the next hour fresh fecal samples were collected and immediately stored at 4°C with long term storage at −70°C.
DNA Extraction, PCR Amplify 16S rRNA, amplicon sequence and sequence data processing
Microbial genome DNA was extracted from fecal samples using QIAamp DNA stool mini kit (QIAGEN, cat#51504) following the manufacture's recommendation. The V3, V4, V1–V3 and V4–V6 hypervariable regions of 16S rRNA were PCR amplified from microbial genome DNA which were harvested from fecal samples using barcoded fusion primers (forward primers: 5′ACTCCTACGGGAGGCAGCAG3′, 5′AYTGGGYDTAAAGNG 3′, 5′AGAGTTTGATCCTGGCTCAG3′ and 5′GTGCCAGCMGCNGCGG3′, reverse primers: 5′TTACCGCGGCTGCTGGCAC3′, 5′TACNVGGGTATCTAATCC 3′, 5′TTACCGCGGCTGCTGGCAC3′, 5′GGGTTNCGNTCGTTG3′ for V3, V4, V1–V3 and V4–V6, respectively). The annealing temperature and extend time are 63.8°C, 30 sec, 42.3°C, 30 sec, 55°C, 60 sec and 55°C, 60 sec for V3, V4, V1–V3 and V4–V6, respectively. The PCR condition were 94°C for 5 min; 94°C for 30 sec, annealing temperature for 30 sec and 72°C for extend time, repeat for 30 cycles; 72°C 7 min. PCR product was excised from a 1.5% agarose gel and purified by QIAquick Gel Extraction Kit (QIAGEN, cat# 28706). Barcoded V4–V6 and V1–V3 amplicons were sequenced by Roche GS FLX+. Barcoded V3 and V4 amplicons were sequenced using pair-end method by Illumina Miseq with a 6 cycle index read. Sequences with an average phred score lower than 25, containing ambiguous bases, homopolymer run exceeds 6, having mismatches in primers, or sequence length shorter than 100 bp were removed. For V3 and V4 pair-end reads, only sequences that overlap longer than 10 bp and without any mismatch were assembled according to their overlap sequence. Reads which could not be assembled were discarded. Barcode and sequencing primers were trimmed from sequence (V1–V3 and V4–V6) or assembled sequence (V3 and V4). Trimmed sequences were uploaded to MGRAST for further study.
Taxonomy classification and statistical analysis
Each sample's trimmed sequence was compared to the RDP, Greengene and SSU databases using the best hit classification option to classify the abundance count of each taxon. The classification parameters were 8 for maximum e-value cutoff, 98 for minimum % identity cutoff and 120 bp for minimum alignment length cutoff. This process was archived by MGRAST15. The metagenome sequences used in this paper are publicly available from the MGRAST (http://metagenomics.anl.gov/).
The abundance count was transformed by log2 and then normalized as follows: From each log transformed measure, the arithmetic mean of all transformed values was subtracted and the difference divided by the standard deviation of all log transformed values for the given sample. After this procedure, the abundance profiles for all samples will exhibit a mean of 0 and a standard deviation of 1. ANOVA was performed by stats package in R following the model of Y = genotype + gender + genotype by gender + error. T-tests were perform by Microsoft excel and all p-values were adjusted by FDR using the BH method by mt.rawp2adjp function in R (http://faculty.mssm.edu/gey01/multtest/multtest-manual.pdf). Heritability and its estimation accuracy were calculated using AI-REML algorithm by wombat (didgeridoo.une.edu.au/km/wombat.php).
References
Spor, A., Koren, O. & Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nature reviews. Microbiology 9, 279–290 (2011).
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108 (2011).
Wen, L. et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455, 1109–1113 (2008).
Ley, R. E. et al. Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America 102, 11070–11075 (2005).
Benson, A. K. et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proceedings of the National Academy of Sciences of the United States of America 107, 18933–18938 (2010).
Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009).
Tims, S., Zoetendal, E. G., de Vos, W. M. & Kleerebezem, M. Metagenomics of the Human Body. (Springer, Berlin; 2011).
Rawls, J. F., Mahowald, M. A., Ley, R. E. & Gordon, J. I. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 127, 423–433 (2006).
Khachatryan, Z. A. et al. Predominant role of host genetics in controlling the composition of gut microbiota. PloS one 3, e3064 (2008).
Salzman, N. H. et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nature immunology 11, 76–83 (2010).
Vijay-Kumar, M. et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328, 228–231 (2010).
Dunnington, E. A. & Siegel, P. B. Long-term divergent selection for eight-week body weight in white Plymouth rock chickens. Poultry science 75, 1168–1179 (1996).
Marquez, G. C., Siegel, P. B. & Lewis, R. M. Genetic diversity and population structure in lines of chickens divergently selected for high and low 8-week body weight. Poultry science 89, 2580–2588 (2010).
Jacobsson, L. et al. Many QTLs with minor additive effects are associated with a large difference in growth between two selection lines in chickens. Genetical research 86, 115–125 (2005).
Meyer, F. et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC bioinformatics 9, 386 (2008).
Youssef, N. et al. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Applied and environmental microbiology 75, 5227–5236 (2009).
Ridlon, J. M., Kang, D. J. & Hylemon, P. B. Bile salt biotransformations by human intestinal bacteria. Journal of lipid research 47, 241–259 (2006).
Nicholson, J. K. et al. Host-gut microbiota metabolic interactions. Science 336, 1262–1267 (2012).
Ley, R. E., Turnbaugh, P. J., Klein, S. & Gordon, J. I. Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023 (2006).
Groh, H., Schade, K. & Horhold-Schubert, C. Steroid metabolism with intestinal microorganisms. Journal of basic microbiology 33, 59–72 (1993).
Jones, B. V., Begley, M., Hill, C., Gahan, C. G. & Marchesi, J. R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proceedings of the National Academy of Sciences of the United States of America 105, 13580–13585 (2008).
Swann, J. R. et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proceedings of the National Academy of Sciences of the United States of America 108 Suppl 1, 4523–4530 (2011).
Lord, R. S. & Bralley, J. A. Clinical applications of urinary organic acids. Part 2. Dysbiosis markers. Alternative medicine review : a journal of clinical therapeutic 13, 292–306 (2008).
Zheng, X. et al. The footprints of gut microbial-mammalian co-metabolism. Journal of proteome research 10, 5512–5522 (2011).
Cani, P. D. et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772 (2007).
Serino, M. et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 61, 543–553 (2012).
Meyer, K. WOMBAT: a tool for mixed model analyses in quantitative genetics by restricted maximum likelihood (REML). Journal of Zhejiang University. Science. B 8, 815–821 (2007).
Li, H. et al. Expression profiling of the solute carrier gene family in chicken intestine from the late embryonic to early post-hatch stages. Animal genetics 39, 407–424 (2008).
Choe, D. W., Loh, T. C., Foo, H. L., Hair-Bejo, M. & Awis, Q. S. Egg production, faecal pH and microbial population, small intestine morphology and plasma and yolk cholesterol in laying hens given liquid metabolites produced by Lactobacillus plantarum strains. British poultry science 53, 106–115 (2012).
Avella, M. A. et al. Lactobacillus rhamnosus Accelerates Zebrafish Backbone Calcification and Gonadal Differentiation through Effects on the GnRH and IGF Systems. PloS one 7, e45572 (2012).
Meyer, B., Bessei, A. W., Vahjen, W., Zentek, J. & Harlander-Matauschek, A. Dietary inclusion of feathers affects intestinal microbiota and microbial metabolites in growing Leghorn-type chickens. Poultry science 91, 1506–1513 (2012).
Acknowledgements
This study was supported by the National High Technology Research and Development Program of China 2011AA100901, the Chinese National Science Fund for Distinguished Young Scholars 31125021. Yan Zhang's contribution has been funded in part with federal funds from the National Institute of Allergy and Infectious Diseases under contract no. HHSN272200900040C awarded to B.W. Sobral. The HW and LW chickens are from a colony maintained at Virginia Tech. We thank Christa Honaker for assistance with preparing information presented in figure 1.
Author information
Authors and Affiliations
Contributions
W.C., H.M., P.S. and Y.Z. contributed to the initial design of this project. H.M., P.S., H.W. and Y.Z. collected samples. Z.S., F.T., G.W., H.W., H.Z., J.Z., L.Z., W.Z. and Z.Z. conducted the experiment and next generation sequence with guidance from W.C. and H.M. C.H., L.Z. and Y.Z. conducted bioinformatics analyses. H.M., P.S. and Y.Z. prepared the manuscript of this publication.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
supplement
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Zhao, L., Wang, G., Siegel, P. et al. Quantitative Genetic Background of the Host Influences Gut Microbiomes in Chickens. Sci Rep 3, 1163 (2013). https://doi.org/10.1038/srep01163
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01163
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
