
Abstract
Hot pepper (Capsicum annuum), one of the oldest domesticated crops in the Americas, is the most widely grown spice crop in the world. We report whole-genome sequencing and assembly of the hot pepper (Mexican landrace of Capsicum annuum cv. CM334) at 186.6× coverage. We also report resequencing of two cultivated peppers and de novo sequencing of the wild species Capsicum chinense. The genome size of the hot pepper was approximately fourfold larger than that of its close relative tomato, and the genome showed an accumulation of Gypsy and Caulimoviridae family elements. Integrative genomic and transcriptomic analyses suggested that change in gene expression and neofunctionalization of capsaicin synthase have shaped capsaicinoid biosynthesis. We found differential molecular patterns of ripening regulators and ethylene synthesis in hot pepper and tomato. The reference genome will serve as a platform for improving the nutritional and medicinal values of Capsicum species.
Similar content being viewed by others

Main
Hot pepper is a member of the Solanaceae family. It is a diploid, facultative, self-pollinating crop and is closely related to potato, tomato, eggplant, tobacco and petunia. Solanaceae plants belong to the asterid clade of eudicots, which includes more than 3,000 diverse species worldwide. Many members of the Solanacea family have the same number of chromosomes (n = 12) yet differ drastically in genome size. Hot pepper is one of the oldest domesticated crops in the Western Hemisphere1, is the most widely grown spice in the world and is a major ingredient in most global cuisines2. Hot pepper has a wide variety of uses, including in pharmaceuticals, natural coloring agents and cosmetics, as an ornamental plant and as the active ingredient in most defense repellents. Hot pepper provides many essential vitamins, minerals and nutrients that have great importance for human health3,4,5,6. In 2011, the top 20 pepper-producing countries grew 33.3 million tons of hot pepper planted on 3.8 Mha (United Nations Food and Agriculture Organization (FAO) statistics; see URLs). In the last decade, world production of hot pepper increased by 40%.
The pungency of hot pepper is due to the accumulation of capsaicinoids, a group of alkaloids that are unique to the Capsicum genus. The heat sensation created by these capsaicinoids is such a defining aspect of this crop that the genus name Capsicum comes from the Greek kapto, which means 'to bite'. Capsaicin, dihydrocapsaicin and nordihydrocapsaicin constitute the primary capsaicinoids, which are produced exclusively in glands on the placenta of the fruit. The organoleptic sensation of heat caused when capsaicin binds to the mammalian transient receptor potential vanilloid 1 (TRPV1) receptor in the pain pathway7 can be argued to be a sixth taste along with sweet, sour, bitter, salty and umami (savory). Many enzymes involved in capsaicinoid biosynthesis are not well characterized, and regulation of the pathway is not fully understood. With more than 22 capsaicinoids isolated from hot pepper, this genus provides an excellent example for exploring the evolution of secondary metabolites in plants2. Capsaicinoids have been found in nature to have antifungal and antibacterial properties, to act as a deterrent to animal predation when ingested and to have inherent properties that aid in avian seed dispersal. Capsaicinoids have many health benefits for humans: they are effective at inhibiting the growth of several forms of cancer8,9,10, are an analgesic for arthritis and other pain11, reduce appetite and promote weight loss12,13,14. It is surprising that a complete understanding of the capsaicinoid pathway at the molecular level is lacking, considering the economic and cultural importance of capsaicinoids.
Here we report a high-quality genome sequence for hot pepper. C. annuum cv. CM334 (Criollo de Morelos 334), a landrace collected from the Mexican state of Morelos, has consistently exhibited high levels of resistance to diverse pathogens, including Phytophthora capsici, pepper mottle virus and root-knot nematodes. This landrace has been extensively used in hot pepper research and cultivar breeding. We also provide resequencing data for two cultivated peppers and for a wild species, C. chinense. Comparative genomics of members of the Solanaceae family, which includes hot pepper, provides an evolutionary view into the genome expansion, origin of pungency, distinct ripening process and disease resistance of hot pepper. This high-quality reference genome of hot pepper will serve as a platform for improving the horticultural, nutritional and medicinal values of Capsicum species.
Results
Sequencing, assembly and genetic variation
We generated 650.2 Gb (186.6× genome coverage) of whole-genome shotgun sequence from C. annuum cv. CM334 (hereafter, CM334) by Illumina sequencing of genomic libraries with insert sizes ranging from 180 bp to 20 kb (Supplementary Figs. 1, 2, 3, 4, 5, 6, Supplementary Tables 1–5 and Supplementary Note). On the basis of 19-mer analysis, we estimated the size of the genome to be 3.48 Gb (Supplementary Fig. 2). For each library, we confirmed that raw data were unbiased by measuring the distribution of insert sizes (Supplementary Fig. 3). After filtering, we assembled 3.06 Gb (87.9% of the 3.48-Gb total) into 37,989 scaffolds (N50 = 2.47 Mb) using SOAPdenovo15 and SSPACE16, and 90% of the genome assembly was contained in 1,276 scaffolds (Table 1 and Supplementary Tables 3 and 4). We validated the genome assembly using 27 BAC sequences from CM334: 26 BAC sequences were fully covered by a single or multiple scaffolds and showed identities of greater than 99.9% (Supplementary Fig. 4 and Supplementary Table 5). To construct pseudomolecules, we established a high-density genetic map with 6,281 markers using 120 recombinant inbred lines derived from C. annuum cv. Perennial and C. annuum cv. Dempsey (hereafter, Perennial and Dempsey) (Supplementary Tables 6–9 and Supplementary Note). We anchored scaffolds to the high-density genetic map (4,562 markers) and to the previously reported genetic map17. Overall, we anchored 86.0% of the assembly (2.63 Gb; 1,357 scaffolds) as 12 chromosome pseudomolecules and ordered them (75.6%; 1,048 scaffolds) on the basis of genetic distance (Supplementary Fig. 7 and Supplementary Table 8).
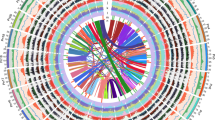
We performed resequencing of two pepper cultivars (Perennial and Dempsey) and de novo sequencing of a wild species (C. chinense PI159236; hereafter, C. chinense) to provide a comprehensive overview of genetic variation and differences in genome structure among pepper cultivars (Supplementary Figs. 8 and 9, Supplementary Tables 2 and 10–19, and Supplementary Note). The proportion of the genome that was divergent between CM334 and the three other pepper genomes was 0.35, 0.39 and 1.85% (10.9, 11.9 and 56.6 million SNPs for Perennial, Dempsey and C. chinense, respectively) (Supplementary Table 11). Divergent sequences were widely dispersed along the pepper chromosomes (Fig. 1 and Supplementary Tables 12 and 13). The number of low-coverage blocks (190 with 500-kb windows) that were divergent between C. annuum and C. chinense shows the genomic variation in the two species (Fig. 1 and Supplementary Table 16).

Left to right: density of matched blocks, gene density, repeat coverage and SNP density. Density of matched blocks is presented for C. chinense, Dempsey and Perennial (left to right) for 500-kb windows. Gene density is presented as the number of genes within 1-Mb intervals. Coverage by repeats represents the proportion of total TEs, Gypsy elements and Copia elements of LTRs within 1-Mb intervals. SNP density is presented as the number of SNPs per 1-Mb interval.
Transposable elements (TEs) have multiple roles in driving genome evolution in eukaryotes18. In total, we identified 2.34 and 2.35 Gb (76.4 and 79.6%, respectively) of sequence in the assembled CM334 and C. chinense genomes as TEs (Table 1 and Supplementary Table 20). The predominant type of TE was long terminal repeat (LTR) elements, which represented approximately 1.7 Gb (more than 70%) of the total number of TEs in the two genomes. Most of the LTRs were Gypsy elements, which accounted for 67.0 and 62.1% of TEs in CM334 and C. chinense, respectively. A large number of Caulimoviridae elements were unique to either pepper genome (Supplementary Table 20). The TEs were widely dispersed throughout the pepper genome and often led to the conversion of euchromatin into heterochromatin. The distribution of TEs was inversely correlated with gene density (Fig. 1).
Gene prediction, gene annotation and RNA sequencing
A total of 34,903 protein-coding genes were predicted in the PGA pipeline (Pepper Genome Annotation v. 1.5) (Supplementary Figs. 10, 11, 12, Supplementary Tables 21–28 and Supplementary Note). This gene number is approximately the same as for tomato (International Tomato Annotation Group (iTAG) v2.3; 34,771 genes)19 and potato (Potato Genome Sequencing Consortium (PGSC) v3.4; 39,031 genes)20, which suggests a similar gene number in Solanaceae plants (Supplementary Figs. 13 and 14). We evaluated consensus gene models using 19.8 Gb of Illumina RNA sequencing (RNA-seq) data. Overall, 93.2% of the predicted coding sequences were supported by Illumina data, demonstrating the high accuracy of gene prediction by PGA. To validate and improve gene models, we manually curated inaccurately annotated genes: 335 genes were manually added, and 86 genes were reclassified as pseudogenes. This manual inspection and curation resulted in the replacement of 1,789 genes with better gene models.
We performed genome-wide analysis of small RNAs and identified 177 microRNAs corresponding to 37 microRNA families (Supplementary Table 26). The distribution of small RNAs correlated well with gene density in the hot pepper genome (Supplementary Fig. 11), similar to in tomato20 but in contrast to what is observed in Arabidopsis thaliana.
In total, we identified 17,397 orthologous gene sets by comparison of the pepper and tomato genomes. To compare gene expression in the pepper and tomato genomes, we performed RNA-seq analyses of the placenta and pericarp at seven crucial stages of fruit development and compared gene expression in other tissues from these two species (Supplementary Fig. 10 and Supplementary Table 22). This tissue-by-tissue analysis showed that a significant change in gene expression patterns of orthologous genes (adjusted P value < 0.01) occurred in 8.8% of the orthologous gene sets in leaf tissue and in 46.4% of the orthologous gene sets in pericarp tissue at 35 d post-anthesis (d.p.a.) (Supplementary Fig. 15).
Genome expansion
The hot pepper genome shared highly conserved syntenic blocks with the genome of tomato, its closest relative within the Solanaceae family (Fig. 2a and Supplementary Fig. 16). However, the hot pepper genome was approximately fourfold larger than the tomato genome, owing to a greater accumulation of repetitive sequences in both heterochromatic and euchromatic regions (Fig. 2b and Supplementary Fig. 17). The most common repeats in the hot pepper genome were LTR retrotransposons, as in many other plant genomes18,21,22,23. However, the composition of LTR retrotransposons in the hot pepper genome was distinct from that for other plants. We estimated the total number of LTR retrotransposons by counting the reverse-transcriptase (RT) domains encoded by the hot pepper and tomato genomes (Fig. 2c). Of the RT domains encoded by the hot pepper genome, there were 12-fold more from the Gypsy family than from the Copia family, in contrast to the relative numbers observed for other plant genomes such as tomato, maize and barley19,21,22. Therefore, substantial proliferation of the Gypsy family was the main cause of expansion of the hot pepper genome.

(a) Circular diagram showing genetic collinearity between hot pepper and tomato. Hot pepper and corresponding tomato chromosomes are represented by red and blue bars, respectively. Lines link the positions of orthologous gene sets, with line color representing each chromosome set. (b) Top, linear comparison of chromosome 2 in hot pepper (upper bar) and tomato (lower bar). The positions of orthologous gene sets are indicated by lines linking the two bars. Blue and white blocks on the bars indicate repeat and genic regions, respectively. Bottom, comparisons of magnified heterochromatic and euchromatic regions in hot pepper and tomato. The positions of the magnified heterochromatic (α, α′) and euchromatic (β, β′) regions are indicated above. (c) Comparison of copy numbers for Gypsy, Copia and Caulimoviridae elements. The fraction of each repeat element is indicated in parentheses. (d) Distribution of repeat elements on chromosome 10 in hot pepper and tomato. The graphs above and below each bar show repeat and gene densities, respectively. Data are shown for three subgroups of Gypsy elements, Copia and Caulimoviridae in hot pepper and for all subgroups in tomato. (e) Histogram of hot pepper and tomato LTR retrotransposon insertions. The insertion patterns of Gypsy and Copia elements in each species are shown. The vertical gray bar indicates speciation time (19.1 million years ago). (f) Comparison of Caulimoviridae element composition. Phylogenetic trees are shown for hot pepper and tomato Caulimoviridae elements. Common elements and those existing in only one species are depicted by filled and empty triangles, respectively. The null subgroup is depicted by lines.
Of the Gypsy family elements, 83.5% were from the Del subgroup, and these elements accumulated primarily in heterochromatic regions of the hot pepper genome (Fig. 2d and Supplementary Figs. 18 and 19). Del elements are known to selectively accumulate in heterochromatic regions owing to the function of the encoded chromodomain24. However, we often found these Del elements in the collinear regions of the hot pepper genome that correlated with tomato euchromatin, with the insertion of these elements resulting in the formation of heterochromatic gene islands in the hot pepper genome (Fig. 2b). The insertion pattern of Del elements may indicate that the hot pepper genome expanded by increasing the size of the existing heterochromatin and converting euchromatin into heterochromatin. We also observed that the Tat subgroup of the Gypsy family had selectively accumulated in euchromatic regions (Fig. 2d). The accumulation of Copia and Tat elements resulted in the expansion of hot pepper euchromatin.
We estimated the times of insertion for Gypsy and Copia elements using the method described by SanMiguel et al.25 (Fig. 2e and Supplementary Fig. 20). The speciation time of pepper and tomato was reported as 19.1 million years ago26. Speciation time can be estimated from the peak value in frequency analysis of the synonymous substitution rate (Ks) of orthologous gene sets27. Therefore, we analyzed a histogram of Ks values from 17,397 orthologous gene sets in hot pepper and tomato. The peak value of the Ks frequency used to determined the speciation time point was observed at 0.3 (19.1 million years ago) (Supplementary Fig. 20). Gypsy elements in the hot pepper genome were gradually accumulated before speciation and peaked in frequency at a substitution value of 0.2 (12.7 million years ago) (Fig. 2e). Copia elements showed relatively recent insertion during the period corresponding to substitution values of between 0 and 0.2, which coincides with the insertion of Gypsy and Copia elements in the tomato genome (Fig. 2e). Variations in heterochromatin can create species barriers28. Thus, the unequal accumulation of Gypsy elements in heterochromatic regions of the progenitor species may have had a role in the speciation of hot pepper.
Among the RT domains encoded by the hot pepper genome, the RT domains of Caulimoviridae were unusually abundant (4.9%) (Supplementary Fig. 21). The number of Caulimoviridae RT domains in hot pepper was 4,304, 9.2-fold more than that observed in tomato. Caulimoviridae is a DNA pararetrovirus of ∼8-kb unit length that evolved from a Gypsy element and replicates via an RNA intermediate without LTR sequences29. So far, Caulimoviridae elements have not been reported in repeat classification in other plant genome sequences, except for a small copy number in the banana genome30. We identified three subgroups of Caulimoviridae including Petuvirus, Caulimovirus and Cavemovirus in the hot pepper genome, but only Cavemovirus was identified in the tomato genome (Fig. 2f). This finding indicates that the proliferation of Petuvirus and Caulimovirus elements resulted in the high abundance of Caulimoviridae in the hot pepper genome with random distribution (Fig. 2d and Supplementary Fig. 19). Therefore, the accumulation of these elements might also have had a role in the expansion of the hot pepper genome in both heterochromatic and euchromatic regions.
Evolution of the capsaicin biosynthetic pathway
Capsaicinoids are the determinants of pepper pungency. They are specialized secondary metabolites found only in Capsicum species. Capsaicinoids are synthesized by capsaicin synthase (CS and Pun1), which condenses vanillylamine from the phenylpropanoid pathway with 8-methyl-6-nonenoyl-CoA from the branched-chain fatty-acid pathway31,32 (Fig. 3a). Although the biosynthetic genes have been partly elucidated33,34,35, the biochemical reactions, evolution and regulation of capsaicinoid biosynthesis are still largely unknown.

(a) Capsaicinoid biosynthetic pathway. Named enzymes marked by red arrows correspond with the genes used in the analysis. (b) Comparison of transcriptional profiles for capsaicinoid biosynthetic genes. Heat maps show log2-scaled reads per kilobase per million reads (RPKM) for biosynthetic genes in CM334 (pungent pepper) and ECW (non-pungent pepper) and for their tomato orthologs. Tissues synthesizing capsaicinoid are indicated by asterisks. PAL, phenylalanine ammonia-lyase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumaroyl-CoA ligase; HCT, hydroxycinnamoyl transferase; C3H, p-coumaroyl shikimate/quinate 3-hydroxylase; COMT, caffeoyl-CoA 3-O-methyltransferase; HCHL, hydroxycinnamoyl-CoA hydratase lyase; AMT, aminotransferase; BCAT, branched-chain amino acid aminotransferase; Kas, ketoacyl-ACP synthase; ACL, acyl carrier protein; FatA, acyl-ACP thioesterase; CS, capsaicin synthase. Three biological replicates from pooled tissues were prepared for RNA-seq. (c) Microsynteny analysis of the hot pepper sequence containing CS (encoding capsaicin synthase; upper bar) and its collinear tomato sequence (lower bar). Lines linking the two bars indicate regions with >70% similarity. CS paralogs and their corresponding genes in tomato are marked by arrows. Numbers above the arrows and letters below the arrows indicate multiplied paralogs. Black and red arrows indicate different origins for the paralogs. (d,e) Models of multiple gene duplications for CS paralogs (d) and their corresponding genes in tomato (e). Branch length in each phylogenetic tree is proportional to the synonymous substitution rate. The vertical gray bar on each tree indicates speciation time. α and β indicate the ancestral genes of the paralogs. β with serial numbers indicate duplicated ancestral genes. Reconstructed duplication events for the paralogs are shown to the right of each tree. Black and red boxes indicate different origins for the paralogs. Solid and dashed lines indicate duplication and translocation events, respectively.
Using homology, microsynteny and previous reports35, we identified all orthologous genes of the capsaicinoid pathway in the tomato genome (Supplementary Fig. 22). In a comparative transcriptome analysis, several genes in the pathway clearly showed differential expression in pepper and tomato fruits (Fig. 3b, Supplementary Fig. 23 and Supplementary Tables 29–31). Fruit-specific expression of CS, encoding a homolog of acyltransferase, primarily occurred during pepper placenta development (at 16 d.p.a., 25 d.p.a. and mature green (MG)). All other genes in the pathway were also expressed at this stage, and capsaicinoids were synthesized in the placenta throughout this period36. In contrast, the orthologous genes in the tomato pathway (BCAT, Kas and CS) were rarely expressed at this stage, and we obtained a similar result for the potato genome (Supplementary Fig. 24 and Supplementary Tables 32 and 33). These results may indicate that changes in the gene expression of BCAT, Kas and CS enabled capsaicinoid synthesis in hot pepper fruits.
Genome-wide or local gene duplication is crucial for the origin of new gene functions37. Microsynteny analysis of the genomic regions surrounding CS in hot pepper (∼436 kb) and tomato (∼183 kb) identified acyltransferase gene clusters in both species (Fig. 3c). Phylogenetic analysis of the acyltransferase gene family within these regions in hot pepper (seven copies) and tomato (four copies) showed that CS appeared after speciation through multiple gene duplications. The seven copies of CS in hot pepper underwent five rounds of unequal tandem duplication events, whereas the four copies of CS in tomato experienced two rounds of duplication events from the ancestral genes (Fig. 3d,e). CS likely emerged only after the final round of gene duplication in the hot pepper genome. Two other genes (Kas and COMT) in the capsaicinoid biosynthetic pathway also underwent unequal gene duplication events similar to those for the orthologous genes in tomato (Supplementary Fig. 22). The biochemical functions of the acyltransferases within both clusters have not been addressed; however, it seems that neofunctionalization occurred with respect to both gene expression and protein function, conferring a role for CS in capsaicinoid synthesis after recent gene duplication. These results provide substantial new insight into the origin of pungency in hot pepper.
We compared expression of the capsaicinoid biosynthetic genes in the placentas of pungent and non-pungent peppers. Non-pungent peppers have a large deletion in CS that spans the region from the promoter to the first exon33. During placenta development, CS was highly expressed only in pungent pepper and was barely expressed in non-pungent pepper (Fig. 3b). All other genes in the capsaicinoid biosynthetic pathway showed similar expression, except for BCAT, COMT and FatA at 6 d.p.a. This result indicates that non-pungent pepper species appeared because of loss of CS expression without substantial changes in the expression of other genes in the biosynthetic pathway.
Gene family analysis
The distribution of orthologous gene families in hot pepper, tomato, potato, Arabidopsis, grape and rice was defined using OrthoMCL38. We identified 23,245 hot pepper genes in 16,345 families, with 7,826 families shared by all 6 species (Supplementary Fig. 25, Supplementary Tables 34–37 and Supplementary Note). A total of 2,139 gene families were unique to Solanaceae plants, and 756 gene families were unique to hot pepper. The hot pepper genome shared 27, 51 and 20 gene families with Arabidopsis, grape and rice, respectively. Variations in family size were found in many hot pepper gene families. We found that gene families involved in disease resistance and cellular functions, such as cytochrome P450 and heat shock protein 70 genes, were significantly expanded in the hot pepper genome (Supplementary Figs. 26–45, Supplementary Tables 38–52 and Supplementary Note).
We identified 2,153 transcription factors (6.25% of the total genes) and transcriptional regulators in 80 gene families. Some transcription factors included Solanaceae-specific subclasses, specifically in the ARF, AP2/ERF, WRKY and NAC families. These transcription factors may have unique functions in Solanaceae, such as defense responses. Nine transcription factor families had fewer genes (including the AP2/ERF family) compared with other plant genomes, and no transcription factor of the DBP family was found in the hot pepper genome (Supplementary Table 43).
A total of 684 genes from the nucleotide-binding site–leucine-rich repeat (NBS-LRR) family were significantly expanded in the pepper genome compared with the other plant genomes (Supplementary Tables 38, 39 and 41). NBS-LRR proteins are identified primarily as disease-resistance genes39. The hot pepper genome contained 636 non-TIR (Toll/interleukin-1 receptor)-type NBS-LRRs, a number significantly higher than the 525 non-TIR NBS-LRRs in rice40. The number of TIR-type proteins in the hot pepper genome (48) was similar to that in potato (47) (Supplementary Table 39). More than half of the NBS-LRR subclasses in each Solanaceae genome were classified into 37 subclasses (Supplementary Table 41). Notably, the Bs2 (bacterial spot resistance gene)41-containing subclass (82 genes) exhibited explosive expansion in the hot pepper genome compared to the tomato (3 genes) and potato (1 gene) genomes. This expansion might be a consequence of evolutionary events of tandem duplication resulting in preferential clustering of the genes on chromosome 9 (Supplementary Fig. 26 and Supplementary Table 42). Expansion of NBS-coding genes in the hot pepper genome resulted in the loss of collinearity with tomato or potato in NBS-coding regions, whereas higher synteny was maintained between the NBS-coding regions of tomato and potato (Supplementary Fig. 27). Comparisons of hot pepper R genes among Solanaceae plants suggested that expansion and diversification of R genes have been involved in lineage-specific parallel evolution through unequal gene-duplication events, resulting in different gene repertoires even in closely related species.
Comparative fruit ripening
Fleshy fruits are physiologically classified into two groups: climacteric and non-climacteric. Climacteric fruits such as tomato and banana display increases in respiration rate and ethylene synthesis during ripening. Non-climacteric fruits such as pepper and strawberry exhibit neither a respiratory burst nor elevated ethylene production during ripening42. Thus, pepper and tomato provide suitable models for comparisons of fruit ripening processes. Gene repertories related to fruit ripening in hot pepper and tomato are well conserved (Supplementary Table 53), which suggests that a gene regulatory mechanism likely causes differentiation in fruit ripening. To identify conserved and differential regulatory mechanisms in hot pepper and tomato, we investigated orthologous regulatory genes previously identified in tomato ripening. Expression of transcription factor genes (RIN43, TAGL1 (ref. 44) and NOR45) and genes involved in ethylene signaling pathways (NR46, ETR4 (ref. 47), EIN2 (ref. 48) and EIL families49) was conserved during fruit ripening (Fig. 4). In contrast, CNR50, Uniform (Golden-like 2)51 and HB-1 (ref. 52) showed distinct expression patterns in hot pepper and tomato (Fig. 4). CNR was expressed at very low levels during pepper ripening, whereas it was expressed at high levels during tomato ripening. The major ethylene biosynthetic genes for tomato ripening, including ACS2, ACS4 and ACO1 (ref. 53), were expressed at very low levels during hot pepper ripening (Fig. 4). Thus, the conservation and divergence of the transcription of these genes and their interactions may lead to qualitative and quantitative differences in the physiological phenomena underlying ripening.

(a) Heat map of normalized RNA-seq data prepared from three biological replicates for genes involved in fruit ripening. SPL-CNR, SQUAMOSA promoter-binding protein-like–colorless non-ripening; GLK2, golden 2-like; HB-1, HD-Zip homeobox protein; ACS, ACC synthase; ACO, ACC oxidase; CCS, capsanthin-capsorubin synthase; CYC-B, chromoplast-specific lycopene β-cyclase; MADS-RIN, MADS-box transcription factor–ripening inhibitor; TAGL1, tomato AGAMOUS-like 1; NAC-NOR, NAC transcription factor–non-ripening; TDR4, tomato FRUITFULL homolog; NR, never ripe; ETR4, ethylene receptor homolog 4; EIN2, ethylene-insensitive; EIL, EIN-like; ERF6, ethylene responsive factor 6; PSY1, phytoene synthase 1. I, divergent gene expression; II, conserved gene expression. (b) Working model of the control of non-climacteric ripening in pepper. Blue and red boxes represent genes that are downregulated and upregulated in pepper, respectively. Orange boxes represent genes that show similar expression in pepper and tomato. Double lines indicate an orthologous relationship between pepper and tomato genes.
The major pigments in pepper fruits are capsanthin and capsorubin, which are pepper-specific carotenoids synthesized by capsanthin-capsorubin synthase (CCS)54. CCS exhibits lycopene β-cyclase activity54 and has an orthologous relationship with chromoplast-specific lycopene β-cyclase (CYC-B)55, which exhibits ethylene-dependent repression44 during tomato ripening. CCS expression was extremely high during pepper ripening (Fig. 4 and Supplementary Table 22), which suggests that ethylene-dependent regulation may be preserved in both types of fruit ripening and lead to distinct outcomes. Therefore, these developmental and hormonal regulatory networks might be the main components that distinguish different ripening patterns.
One of the ripening characteristics distinguishing pepper and tomato is fruit softening, in which polygalacturonase (PG) has a central role. The hot pepper PG gene encoded a partial deletion of ∼90 amino acids in the C-terminal region of the protein compared to tomato PG (LePG2a, Solyc10g080210) (Supplementary Fig. 46). In comparative sequencing analysis of PG (CA10g18920) from wild-type pepper and the Soft flesh56 mutant, we found that a point mutation in the 3′ splice acceptor site at intron VIII generated a premature stop codon in the PG gene from wild-type pepper. The SNP in PG genetically cosegregated with the fruit softening phenotype and distinguished normal and soft-fleshed fruits among pepper germplasms (Supplementary Fig. 47 and Supplementary Table 54). The levels of water-soluble pectin in the red fruit from the Soft flesh mutant were much higher than in the fruit from wild-type pepper (Supplementary Fig. 48). The differential levels of water-soluble pectin likely supported PG-mediated pectin degradation and resultant fruit softening. Therefore, the impaired PG gene in wild-type hot pepper may have a pivotal role in the non-softening of fruit in coordination with transcriptional regulation of cell wall–related genes (Supplementary Table 55).
Ascorbate (vitamin C) is an essential nutrient for humans and acts as an antioxidant57. Pepper fruit is one of the richest sources of ascorbate. The concentration of ascorbate in pepper is up to tenfold higher than in tomato58. Most of the pepper genes in the L-galactose pathway showed expression similar to or higher than in tomato (Supplementary Table 56). GGP1, which catalyzes the committed steps for L-galactose synthesis, was highly expressed in all stages of pepper fruit development compared to in pepper vegetative tissues. The expression of pepper GGP1 was two- to threefold higher during the green-fruit stages (at 6, 16 and 25 d.p.a.) compared to in tomato (Supplementary Fig. 49). These data indicate that the L-galactose pathway may be the predominant biosynthetic pathway for ascorbate in hot pepper. Recycling is another factor that controls ascorbate content59. Ascorbate oxidases (APXs) generate dehydroascorbate; ascorbate can be regenerated by monodehydroascorbate reductase (MDHAR) and dehydroascorbate reductase (DHAR). APX2 expression in tomato breaker fruits was 20-fold higher than in hot pepper. In contrast, DHAR was highly expressed during hot pepper ripening, with the highest expression observed at 16 d.p.a. for pepper fruits, at a level 5-fold higher than in tomato. These differentially expressed genes involved in ascorbate biosynthesis and recycling further explain the greater accumulation of ascorbate in pepper fruit.
Discussion
In 2011, the value of global hot pepper production was $14.4 billion, 40-fold higher than in 1980 (FAO statistics; see URLs). Pepper consumption continues to grow because of this fruit's high nutritional value. The pepper genome sequences described here can serve as an important genomic resource for improving the nutritional and pharmaceutical value derived from hot pepper and for supporting evolutionary and comparative genomic studies of Solanaceae, one of the world's most diversified plant families. Capsicum is the only genus that evolved the biosynthesis of capsaicinoids, which consist of more than 20 related alkaloids that cause pungency in pepper fruit. The hot pepper genome sequence will provide an opportunity to gain a complete understanding of the capsaicinoid pathway and represents an excellent resource for exploring the evolution of secondary metabolites in plants. This study strongly suggests that pepper pungency originated through the evolution of new genes by unequal duplication of existing genes and owing to changes in gene expression in fruits after speciation. The hot pepper genome provides a strong foundation for further studies using comparative genomics, metabolic engineering and transgenic approaches to unveil the complete pathway of capsaicinoid biosynthesis in Capsicum species. In combination with the recently published tomato19 and potato20 genomes, the hot pepper genome will elucidate the evolution, diversification and adaptation of more than 3,000 Solanaceae species, which are adapted to a wide range of geoecological habitats ranging from the driest deserts to tropical rainforests. Resequencing of two cultivars and de novo sequencing of C. chinense provides a landscape of genomic diversity among Capsicum species. The hot pepper genome will enable the advancement of new breeding technologies through the exploration of genome-wide associations and genomic selection studies on horticulturally important traits such as fruit size, yield, pungency, tolerance to abiotic stresses, nutritional content and resistance to multiple diseases.
Methods
De novo and resequencing of pepper genomes.
A Mexican landrace, C. annuum cv. CM334, and a wild species, C. chinense PI159236, were used for de novo genome sequencing, and C. annuum cv. Perennial and C. annuum cv. Dempsey were resequenced. Paired-end and mate-pair libraries for sequencing were prepared with the corresponding kits (Illumina) following the manufacturer's instructions and were validated with KAPA SYBR FAST Master Mix Universal 2× qPCR Master Mix (Kapa Biosystems). Constructed libraries were sequenced on Illumina platforms (Genome Analyzer IIx and HiSeq 2000) using standard protocols (Supplementary Note).
Genome assembly.
Before genome assembly, short-read sequences from each library were preprocessed using in-house preprocessing pipelines to increase the accuracy of genome assembly (Supplementary Note). Contamination from bacterial sequences, duplicated short reads and low-quality bases in each short-read sequence was removed. Preprocessed short reads were error corrected using Quake60. Remaining sequence was then assembled using SOAPdenovo15 with the optimal K-mer for each library (Supplementary Note). The assembled RCM334 genome sequence was validated with 27 BACs with insert size larger than 70 kb from euchromatic or heterochromatic regions (Supplementary Note). The C. chinense genome assembly was assessed using C. chinense ESTs and annotated CM334 genes (Supplementary Note).
Construction of genetic linkage map and pseudomolecules.
A high-density genetic map for hot pepper was constructed with 120 recombinant inbred lines (RILs) derived from an intraspecific cross between Dempsey and Perennial using SNP markers (Supplementary Note). Markers were then aligned to the scaffolds using BLASTN (identity ≥ 98% and coverage ≥ 70%).
Analysis of genomic variations.
Preprocessed raw data for Perennial, Dempsey and C. chinense were mapped to the CM334 reference genome using Bowtie 2 (ref. 61) (Supplementary Note). SAMtools62 was used to call DNA variations. Classification and annotation of DNA variations was performed using SnpEff63.
Transcriptome sequencing and analysis.
CM334 plants were grown under standard conditions (day/night cycles, 27/19 °C, 16/8 h) in a greenhouse. Roots, leaves and stems were harvested from plants 6 weeks after sowing. Pepper pericarp and placenta tissues from CM334, pepper placenta from ECW and tomato placenta from Solanum lycopersicum cv. Alisa Craig were harvested at 6 d.p.a., 16 d.p.a., 25 d.p.a., MG, B, B5 and B10. For transcriptome comparison, previously published RNA-seq data for tomato pericarp was used19. Three biological replicates from pooled tissues were prepared. Total RNA was isolated using TRIzol reagent (Invitrogen). A modified TruSeq method was used to construct a strand-specific RNA-seq library64 with different index primers, and libraries were sequenced on the Illumina HiSeq 2000 system. Resulting reads were aligned to pepper CM334 sequences and tomato Heinz sequences using CLC Assembly Cell (CLC Bio). Counts for mapped reads were normalized by RPKM. Differentially expressed genes during pericarp development were identified using DESeq65 (Supplementary Note).
Genome annotation.
Genome annotation was performed using the PGA pipeline (Supplementary Note). This pipeline uses a combination of evidence-based gene prediction (RNA-seq and proteins) and ab initio gene prediction. Consensus gene models were determined by EVM66, and these models were then updated with PASA assembly alignments. Gene functions were assigned according to the best alignment attained using BLASTP to the UniProt database (including SWISS-PROT and TrEMBL databases) and INTERPRO scan.
Analysis of differential gene expression.
Orthologous gene sets were found by reciprocal BLAST with pepper and tomato coding sequences (Supplementary Note). Analysis of differential gene expression was carried out using DESeq65. Synonymous substitution rates for orthologous gene sets were calculated by codeml in PAML67.
Repeat annotation and genome expansion analysis.
All TE-related repeats were characterized using RepeatMasker with a custom library for pepper. Synonymous substitution rates for LTRs were calculated by codeml in the PAML package67 (Supplementary Note). Visualization of comparative sequence analysis for pepper and tomato was performed with in-house Python scripts or the Circos program68. Phylogenetic trees were constructed using the MEGA5 package69.
OrthoMCL analysis.
Orthologous gene clusters were assigned from OrthoMCL38 with its standard parameters of six species to identify gene families enriched in the hot pepper genome. Gene sets from hot pepper (PGAv1.0), tomato (v2.3), Arabidopsis (TAIR10), grape (VvGDB v2.0), rice (MSU RGAP 7) and potato (PGSC v3.4) were used to infer putative orthologous gene families. Splice variants and incomplete gene models in the genomes were removed, and an all-by-all comparison was then performed using BLASTP with an E value of 1 × 10−5. A total of 161,775 protein sequences were clustered into 21,808 gene families (Supplementary Note).
URLs.
Food and Agriculture Organization of the United Nations (FAO statistics), http://faostat.fao.org/.
Accession codes.
Whole-genome sequences for the pepper have been deposited in GenBank under accession AYRZ00000000 (the version described in the manuscript is the first version, AYRZ01000000). Further information, including the CM334 genome assembly, pseudomolecules, annotations and C. chinense genome assembly are available through our website at http://peppergenome.snu.ac.kr.
Accession codes
References
Aguilar-Meléndez, A., Morrell, P.L., Roose, M.L. & Kim, S.C. Genetic diversity and structure in semiwild and domesticated chiles (Capsicum annuum; Solanaceae) from Mexico. Am. J. Bot. 96, 1190–1202 (2009).
Bosland, P.W.E.J.V.P. Vegetable and Spice Capsicums (CABI, Wallingford, UK, 2012).
Marín, A., Ferreres, F., Tomas-Barberan, F.A. & Gil, M.I. Characterization and quantitation of antioxidant constituents of sweet pepper (Capsicum annuum L.). J. Agric. Food Chem. 52, 3861–3869 (2004).
Matsufuji, H., Ishikawa, K., Nunomura, O., Chino, M. & Takeda, M. Oxidant content of different coloured sweet peppers, white, green, yellow, orange and red (Capsicum annuum L.). Int. J. Food Sci. Technol. 42, 1482–1488 (2007).
Matus, Z., Deli, J. & Szabolcs, J.J. Carotenoid composition of yellow pepper during ripening—isolation of β-cryptoxanthin 5,6-epoxide. J. Agric. Food Chem. 39, 1907–1914 (1991).
Mejia, L.A., Hudson, E., deMejia, E.G. & Vazquez, F. Carotenoid content and vitamin-A activity of some common cultivars of Mexican peppers (Capsicum annuum) as determined by HPLC. J. Food Sci. 53, 1448–1451 (1998).
Caterina, M.J. et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997).
Mori, A. et al. Capsaicin, a component of red peppers, inhibits the growth of androgen-independent, p53 mutant prostate cancer cells. Cancer Res. 66, 3222–3229 (2006).
Surh, Y.J. More than spice: capsaicin in hot chili peppers makes tumor cells commit suicide. J. Natl. Cancer Inst. 94, 1263–1265 (2002).
Ito, K. et al. Induction of apoptosis in leukemic cells by homovanillic acid derivative, capsaicin, through oxidative stress: implication of phosphorylation of p53 at Ser-15 residue by reactive oxygen species. Cancer Res. 64, 1071–1078 (2004).
Fraenkel, L., Bogardus, S.T. Jr., Concato, J. & Wittink, D.R. Treatment options in knee osteoarthritis: the patient's perspective. Arch. Intern. Med. 164, 1299–1304 (2004).
Lejeune, M.P., Kovacs, E.M. & Westerterp-Plantenga, M.S. Effect of capsaicin on substrate oxidation and weight maintenance after modest body-weight loss in human subjects. Br. J. Nutr. 90, 651–659 (2003).
Westerterp-Plantenga, M.S., Smeets, A. & Lejeune, M.P.G. Sensory and gastrointestinal satiety effects of capsaicin on food intake. Int. J. Obes. (Lond.) 29, 682–688 (2005).
Ludy, M.-J., Moore, G.E. & Mattes, R.D. The effects of capsaicin and capsiate on energy balance: critical review and meta-analyses of studies in humans. Chem. Senses 37, 103–121 (2012).
Huang, S. et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41, 1275–1281 (2009).
Boetzer, M., Henkel, C.V., Jansen, H.J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Yarnes, S.C. et al. Identification of QTLs for capsaicinoids, fruit quality, and plant architecture–related traits in an interspecific Capsicum RIL population. Genome 56, 61–74 (2013).
Feschotte, C., Jiang, N. & Wessler, S.R. Plant transposable elements: where genetics meets genomics. Nat. Rev. Genet. 3, 329–341 (2002).
Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Xu, X. et al. Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195 (2011).
Schnable, P.S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115 (2009).
International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716 (2012).
Brenchley, R. et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491, 705–710 (2012).
Gao, X., Hou, Y., Ebina, H., Levin, H.L. & Voytas, D.F. Chromodomains direct integration of retrotransposons to heterochromatin. Genome Res. 18, 359–369 (2008).
SanMiguel, P., Gaut, B.S., Tikhonov, A., Nakajima, Y. & Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 20, 43–45 (1998).
Wang, Y. et al. Euchromatin and pericentromeric heterochromatin: comparative composition in the tomato genome. Genetics 172, 2529–2540 (2006).
Cannon, S.B. et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. USA 103, 14959–14964 (2006).
Hughes, S.E. & Hawley, R.S. Heterochromatin: a rapidly evolving species barrier. PLoS Biol. 7, e1000233 (2009).
Llorens, C. et al. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 39, D70–D74 (2011).
D'Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012).
Bennett, D.J. & Kirby, G.W. Constitution and biosynthesis of capsaicin. J. Chem. Soc. C 1968, 442–446 (1968).
Leete, E. & Louden, M.C.L. Biosynthesis of capsaicin and dihydrocapsaicin in Capsicum frutescens. J. Am. Chem. Soc. 90, 6837–6841 (1968).
Stewart, C. et al. The Pun1 gene for pungency in pepper encodes a putative acyltransferase. Plant J. 42, 675–688 (2005).
del Rosario Abraham-Juárez, M., Carmen Rocha-Granados, M., López, M., Rivera-Bustamante, R. & Ochoa-Alejo, N. Virus-induced silencing of Comt, pAmt and Kas genes results in a reduction of capsaicinoid accumulation in chili pepper fruits. Planta 227, 681–695 (2008).
Mazourek, M. et al. A dynamic interface for capsaicinoid systems biology. Plant Physiol. 150, 1806–1821 (2009).
Fujiwake, H., Suzuki, T. & Iwai, K. Intracellular localization of capsaicin and its analogues in Capsicum fruit. II. The vacuole as the intracellular accumulation site of capsaicinoid in the protoplast of Capsicum fruit. Plant Cell Physiol. 21, 1023–1030 (1980).
Roth, C. et al. Evolution after gene duplication: models, mechanisms, sequences, systems, and organisms. J. Exp. Zool. B Mol. Dev. Evol. 308, 58–73 (2007).
Li, L., Stoeckert, C.J. Jr. & Roos, D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
van Ooijen, G., van den Burg, H.A., Cornelissen, B.J. & Takken, F.L. Structure and function of resistance proteins in solanaceous plants. Annu. Rev. Phytopathol. 45, 43–72 (2007).
Goff, S.A. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100 (2002).
Tai, T.H. et al. Expression of the Bs2 pepper gene confers resistance to bacterial spot disease in tomato. Proc. Natl. Acad. Sci. USA 96, 14153–14158 (1999).
Klee, H.J. & Giovannoni, J.J. Genetics and control of tomato fruit ripening and quality attributes. Annu. Rev. Genet. 45, 41–59 (2011).
Vrebalov, J. et al. A MADS-box gene necessary for fruit ripening at the tomato RIPENING-INHIBITOR (RIN) locus. Science 296, 343–346 (2002).
Vrebalov, J. et al. Fleshy fruit expansion and ripening are regulated by the tomato SHATTERPROOF gene TAGL1. Plant Cell 21, 3041–3062 (2009).
Giovannoni, J. Molecular biology of fruit maturation and ripening. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 725–749 (2001).
Wilkinson, J.Q., Lanahan, M.B., Yen, H.C., Giovannoni, J.J. & Klee, H.J. An ethylene-inducible component of signal transduction encoded by NEVER-RIPE. Science 270, 1807–1809 (1995).
Tieman, D.M., Taylor, M.G., Ciardi, J.A. & Klee, H.J. The tomato ethylene receptors NR and LeETR4 are negative regulators of ethylene response and exhibit functional compensation within a multigene family. Proc. Natl. Acad. Sci. USA 97, 5663–5668 (2000).
Wang, J., Chen, G., Hu, Z. & Chen, X. Cloning and characterization of the EIN2-homology gene LeEIN2 from tomato. DNA Seq. 18, 33–38 (2007).
Tieman, D.M., Ciardi, J.A., Taylor, M.G. & Klee, H.J. Members of the tomato LeEIL (EIN3-like) gene family are functionally redundant and regulate ethylene responses throughout plant development. Plant J. 26, 47–58 (2001).
Manning, K. et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952 (2006).
Powell, A.L. et al. Uniform ripening encodes a Golden 2-like transcription factor regulating tomato fruit chloroplast development. Science 336, 1711–1715 (2012).
Lin, Z. et al. A tomato HD-Zip homeobox protein, LeHB-1, plays an important role in floral organogenesis and ripening. Plant J. 55, 301–310 (2008).
Barry, C.S. et al. Differential expression of the 1-aminocyclopropane-1-carboxylate oxidase gene family of tomato. Plant J. 9, 525–535 (1996).
Hugueney, P. et al. Metabolism of cyclic carotenoids: a model for the alteration of this biosynthetic pathway in Capsicum annuum chromoplasts. Plant J. 8, 417–424 (1995).
Ronen, G., Carmel-Goren, L., Zamir, D. & Hirschberg, J. An alternative pathway to β-carotene formation in plant chromoplasts discovered by map-based cloning of β and old-gold color mutations in tomato. Proc. Natl. Acad. Sci. USA 97, 11102–11107 (2000).
Smith, P.G. Deciduous ripe fruit character in peppers. Proc. Am. Soc. Hort. Sci. 57, 343–344 (1951).
Frei, B., England, L. & Ames, B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. USA 86, 6377–6381 (1989).
Wahyuni, Y., Ballester, A.R., Sudarmonowati, E., Bino, R.J. & Bovy, A.G. Metabolite biodiversity in pepper (Capsicum) fruits of thirty-two diverse accessions: variation in health-related compounds and implications for breeding. Phytochemistry 72, 1358–1370 (2011).
Wang, Z., Xiao, Y., Chen, W., Tang, K. & Zhang, L. Increased vitamin C content accompanied by an enhanced recycling pathway confers oxidative stress tolerance in Arabidopsis. J. Integr. Plant Biol. 52, 400–409 (2010).
Kelley, D.R., Schatz, M.C. & Salzberg, S.L. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 11, R116 (2010).
Langmead, B. & Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Zhong, S. et al. High-throughput Illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 940–949 (2011).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Haas, B.J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Acknowledgements
This work was supported by a grant from the Agricultural Genome Center of the Next-Generation Biogreen21 Program, Rural Development Administration of the Korean government (project number PJ008199012012). S.-I.Y. (Research Fellowship), H.-A.L. (National Junior Research Fellowship) and E.S. (Global Ph.D. Fellowship) were supported by the National Research Foundation (NRF) of the Korean government. The authors are thankful for financial support from the following companies: Hortigenetics, Monsanto, Rijk Zwaan, Syngenta, Semillas Fito, Sakata Seed, Enza Zaden, Nunhems and Takii.
Author information
Authors and Affiliations
Contributions
D.C. conceived the project, designed content and organized the manuscript. Y.D.C., B.-S.P. and Y.-H.L. coordinated the project. S.K., M.P., S.-I.Y., Y.-M.K., J.M.L., H.-A.L., B.-D.K., I.Y., Y.Y., R.W.K., I.-Y.C., B.-S.C., J.-S.L. and E.S. performed data generation and/or analysis and managed subprojects. Y.-M.K., H.-A.L., S.-B.K., H.-Y.L. and S.-Y. Kim prepared DNA and RNA samples. S.K. performed de novo genome sequencing and assembly. Y.-M.K. was genome-annotation coordinator, and E.S., G.-W.L., H.-Y.L., M.-S.K., Y.S., S.-J.N. and J.P. were involved in genome annotation. J.M.L. coordinated comparative fruit ripening analysis, and H.-A.L., J.J.G., B.-D.K. and I.Y. were involved in comparative fruit ripening analysis. M.P. coordinated genome and capsaicinoid biosynthetic pathway analysis. S.K., M.P., S.-I.Y., Y.-M.K., E.S., J.C., K.C., K.-T.K., K.J. and G.-W.L. performed bioinformatics analysis. B.-C.K., Y.D.J., H.-B.Y., H.-J.J., W.-H.K., S.-H.J., B.-W.L., A.v.D., H.A. and T.H. were involved in map construction and development. J.-K.K. performed FISH analysis. M.P., J.M.L., H.-A.L., A.v.D., H.A. and T.H. performed transcriptome data generation and/or analysis. S.-I.Y. coordinated gene family analysis; Y.-M.K., E.S., J.C., S.-K.O., C.B., J.H.H., J.-S.K., O.C., I.P., M.C.S., S.B.L., Y.-K.K., Y.S.S., S.-Y. Kwon, H.A.K., J.M.P., H.-J.K., S.-B.C., H.-T.C., H.-S.C., M.-S.L., C.S., J.Y.L., J.H.P., W.T.K., H.-S.P., H.K.A., J.K.C.R., I.S. and S.-J.L. performed gene family analysis. S.K., M.P., S.-I.Y., Y.-M.K., J.M.L., H.-A.L., E.S., P.W.B., G.R. and D.C. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note, Supplementary Figures 1–49 and Supplementary Tables 1–21, 25–28, 32, 34–36, 38, 39, 41–45, 48, 50–52 and 54 (PDF 12035 kb)
Supplementary Table 22
RPKM table of pepper genes against RNA-seq data of each tissue (XLSX 12544 kb)
Supplementary Table 23
RPKM table of tomato genes against RNA-seq data of placenta tissue (XLSX 4844 kb)
Supplementary Table 24
Differentially upregulated and downregulated genes during ripening (fold change > 2, fold change < 0.05 and adjusted P value < 0.0001) (XLSX 181 kb)
Supplementary Table 29
RPKM values of the genes in the capsaicinoid biosynthetic pathway and their orthologs in tomato (XLSX 12 kb)
Supplementary Table 30
RPKM values of the candidate genes in the capsaicinoid biosynthetic pathway (XLSX 22 kb)
Supplementary Table 31
RPKM values for tomato orthologs of the candidate genes in the capsaicinoid biosynthetic pathway (XLSX 17 kb)
Supplementary Table 33
FPKM values for potato orthologs of the genes in the capsaicinoid biosynthetic pathway (XLSX 9 kb)
Supplementary Table 37
Gene clusters pepper specific and Solaneacea specific (tomato, potato and pepper) identified by orthoMCL (XLSX 209 kb)
Supplementary Table 40
Subgroups of NBS-LRR superfamily between pepper, tomato, potato, Arabidopsis, grape and rice using OrthoMCL (XLSX 17 kb)
Supplementary Table 46
List of predicted CYP450 genes in pepper genome (XLSX 20 kb)
Supplementary Table 47
Gene list and digital expression level of pepper FT family members (XLSX 15 kb)
Supplementary Table 49
List of PPP family (A) and PPM family (B) gene locus numbers and PP2A regulatory subunit (C) from A. thaliana, O. sativa, S. lycopersicum and C. annuum (XLSX 24 kb)
Supplementary Table 53
Gene expression levels and OrthoMCL analysis of ripening-related genes in Figure 4 (XLSX 12 kb)
Supplementary Table 55
Gene expression levels and OrthoMCL analysis of cell wall–related genes in pepper (XLSX 17 kb)
Supplementary Table 56
Gene expression levels and OrthoMCL analysis of ascorbate biosynthetic pathway (XLSX 19 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Kim, S., Park, M., Yeom, SI. et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet 46, 270–278 (2014). https://doi.org/10.1038/ng.2877
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2877
This article is cited by
-
Identification and expression analyses of B3 genes reveal lineage-specific evolution and potential roles of REM genes in pepper
BMC Plant Biology (2024)
-
Genome-wide identification of the B3 transcription factor family in pepper (Capsicum annuum) and expression patterns during fruit ripening
Scientific Reports (2024)
-
Genetics and evolutionary insights from Solanaceae genome sequences
Plant Systematics and Evolution (2024)
-
Characterization of a Homeodomain-Leucine Zipper Gene 12: Gene Silencing in Pepper and Arabidopsis-Based Overexpression During Abiotic Stress
Journal of Plant Growth Regulation (2024)
-
Exogenous strigolactone enhanced the drought tolerance of pepper (Capsicum chinense) by mitigating oxidative damage and altering the antioxidant mechanism
Plant Cell Reports (2024)
