
Abstract
Rhabdoid tumor is characterized by rhabdoid cells and shows complete loss of SMARCB1/INI1 protein expression. In existing classifications, the diagnostic synonyms vary depending on the anatomic site: rhabdoid tumors in the central nervous system or extra-central nervous system are, respectively, classified as atypical teratoid/rhabdoid tumor or malignant rhabdoid tumor. In this study, we analyzed the histological, immunohistochemical, microRNA, and clinicopathological statuses of tumors initially diagnosed as malignant rhabdoid tumor (n=33), atypical teratoid/rhabdoid tumor (n=11), and pediatric undifferentiated/unclassified sarcoma (n=8) with complete loss of SMARCB1/INI1 expression, and considered the possibility of their histological reclassification. Our analysis indicated that the tumors could be histologically reclassified into three groups: conventional-type tumors resembling malignant rhabdoid tumor, atypical teratoid/rhabdoid-type tumors resembling atypical teratoid/rhabdoid tumor, and small cell-type tumors resembling malignant lymphoma. The reclassified conventional type was composed of 27 malignant rhabdoid tumors and 9 atypical teratoid/rhabdoid tumors (36 cases). The atypical teratoid/rhabdoid type consisted of six malignant rhabdoid tumors, two atypical teratoid/rhabdoid tumors, and two undifferentiated/unclassified sarcomas (10 cases). The six cases of small cell type were made up of six undifferentiated/unclassified sarcomas. All of the available tumor specimens were positive for vimentin and epithelial marker (EMA, CAM5.2, or AE1/AE3). MicroRNA profiles were not significantly different between the conventional- and small cell-type tumors (Pearson's correlation coefficient: 0.888300 or 0.891388). There was no significant difference in overall survival between atypical teratoid/rhabdoid tumor and malignant rhabdoid tumor (P=0.16). In addition, there were no significant differences in survival between any of the reclassified combinations. In conclusion, we could classify eight tumors initially diagnosed as undifferentiated/unclassified sarcomas into two cases of atypical teratoid/rhabdoid type and six cases of small cell type. We suggest that reclassification of malignant rhabdoid tumors into three groups according to their histologic features rather than the traditional classification by sites of origin would be favorable for their histopathological diagnosis.
Similar content being viewed by others

Main
The ATP-dependent SWI/SNF complex represents a novel link between chromatin remodeling and tumor suppression, as recurrent mutations in subunits of the complex have now been identified in various cancers.1 SMRRCB1/INI1 is one of the evolutionarily conserved core subunits in this complex and is ubiquitously expressed in the nuclei of all normal cells.2 Disruption of SMARCB1/INI1 expression in mice results in early embryonic lethality: SMARCB1/INI1-null embryos die between 3.5 and 5.5 days postcoitum.3 Loss of SMARCB1/INI1 protein expression has recently been identified in a variety of tumors, and growing evidence indicates that this complex including SMARCB1/INI1 has widespread roles in tumor suppression.1, 2
Malignant rhabdoid tumor was originally described in 1978 as a rhabdomyosarcomatoid variant of Wilms’ tumor; malignant rhabdoid tumor was the first reported SMARCB1/INI1-deficient tumor, and is representative of the others discovered thereafter.4 Following the discovery of malignant rhabdoid tumor, a complete loss of SMARCB1/INI1 protein expression was observed in pediatric neoplasms such as atypical teratoid/rhabdoid tumor, some unclassified sarcomas, and about half of pediatric myoepithelial carcinomas.2, 5, 6, 7 Recently, a rare variant of malignant rhabdoid tumor (SMARCB1/INI1-positive/SMARCA4/BRG1-negative) was also reported.8 These tumors are known to have common histological and immunohistochemical features such as the appearance of rhabdoid cells, which are characterized by the existence of a large eosinophilic inclusion within the cytoplasm, eccentric nuclei and prominent nucleoli, and coimmunoexpression of epithelial and non-epithelial markers.2, 5, 6, 7
In the present study, we analyzed the histological, immunohistochemical, microRNA, and clinicopathological status in tumors initially diagnosed as malignant rhabdoid tumor, atypical teratoid/rhabdoid tumor, and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 expression. Moreover, we proposed the possibility of histological reclassification of these tumors, emphasizing their morphological features.
Materials and methods
Patients
The present cases of malignant rhabdoid tumor, atypical teratoid/rhabdoid tumor, and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 expression were selected from among more than 20 000 cases of bone, soft-tissue, and pediatric tumors registered in the Department of Anatomic Pathology, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan between 1975 and 2015. The primary monoclonal antibody used in the case selection was BAF47, an antibody to the SMARCB1/INI1 gene product (clone 25; 1:250; 20-min microwave; BD Transduction Laboratories, San Diego, CA). Consequently, formalin-fixed, paraffin-embedded specimens of 33 malignant rhabdoid tumors, 11 atypical teratoid/rhabdoid tumors, and 8 pediatric undifferentiated/unclassified sarcomas (no staining of SMARCB1/INI1 in tumor nuclei) (Supplementary Table and Table 1) were available. These samples had been obtained from open biopsy specimens or surgically resected tumors. Case 1 and 2 of Table 1 were previously reported by Hachitanda et al.9 and Tanaka and co-workers,10 respectively. In all cases, the diagnosis was based on light microscopic examination with hematoxylin and eosin (HE) staining according to the most recent World Health Organization classification.5, 11, 12 Each sample was prepared from a different patient.
MicroRNA microarray analysis was performed in six frozen tumor samples (five malignant rhabdoid tumors; one pediatric undifferentiated/unclassified small round cell sarcoma, case 5 of Table 1) and three frozen samples of surrounding non-tumorous skeletal muscle that were collected from patients with various types of sarcoma as controls.
Immunohistochemistry
Immunohistochemical staining using the universal immunoperoxidase polymer method (Envision Kit; Dako-Japan, Tokyo, Japan) or streptavidin–biotin–peroxidase method (Histofine; Nichirei, Tokyo, Japan) was performed for all available cases. Formalin-fixed, paraffin-embedded tissue was sectioned at 3 μm. Antigen retrieval was carried out by boiling the slides with 10 mM sodium citrate (pH 6.0) or Target Retrieval Solution (DAKO, Carpinteria, CA). The primary antibodies and staining conditions are summarized in Table 2. Immunoreactivity was graded semiquantitatively as follows: 0, no staining; 1+, <5% of tumor cells reactive; 2+, 5–25% of tumor cells reactive; 3+, 26–50% of tumor cells reactive; and 4+, >50% of tumor cells reactive. Appropriate controls were used throughout.
MicroRNA Array
Total RNAs, including microRNAs, were extracted from frozen samples using an miRNeasy Mini Kit (Qiagen Japan, Tokyo, Japan) according to the manufacturer’s instructions.
MicroRNA array analysis was conducted in the same way as described previously.13, 14 Extracted total RNA was labeled with Hy5 using a miRCURY LNA Array miR Labeling Kit (Exiqon, Vedbaek, Denmark). Labeled RNAs were hybridized onto 3D-Gene Human miRNA Oligo chips containing 837 antisense probes printed in duplicate spots (Toray, Kamakura, Japan). The annotation and oligonucleotide sequences of the probes were designed to correspond to those in the miRBase miRNA database (http://microrna.sanger.ac.uk/sequences/). The chips were stringently washed, and fluorescent signals were scanned with a ScanArray Lite Scanner (Perkin-Elmer, Waltham, MA) and analyzed using GenePix Pro version 5.0 (Molecular Devices, Sunnyvale, CA). The raw data for each spot were normalized by substituting the mean intensity of the background signal, which was determined from the signal intensities of all the blank spots with 95% confidence intervals. Measurements of both duplicate spots with signal intensities >2 s.d. of the background signal intensity were considered to be valid. The relative expression level of a given miRNA was calculated by comparing the signal intensities of the averaged valid spots with their mean value throughout the microarray experiments after normalization by their median values adjusted equivalently.
Results
Histological Findings
The pediatric SMARCB1/INI1-deficient tumors in the present study, which consisted of malignant rhabdoid tumor, atypical teratoid/rhabdoid tumor, and undifferentiated/unclassified sarcoma, could be classified into three groups based on their histology: conventional type, atypical teratoid/rhabdoid type, and small cell type. The reclassified histological definitions are summarized in Table 3.
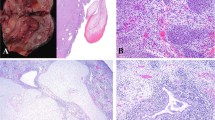
The first type, into which 36 of the present cases were classified (10 of the 14 cases of malignant rhabdoid tumor of the kidney; 17 of the 19 cases of extrarenal malignant rhabdoid tumor; and 9 of the 11 cases of atypical teratoid/rhabdoid tumor) resembled conventional malignant rhabdoid tumor. This conventional type showed a proliferation of epithelioid tumor cells having large, vesicular, and oval-to-polygonal nuclei, prominent nucleoli, and scant-to-abundant eosinophilic cytoplasms, arranged in sheets or in solid trabecular or solid alveolar patterns (Figures 1a and b). Rhabdoid cells with globoid and hyaline cytoplasmic inclusion and eccentric nuclei were focally-to-frequently recognized. Tumor necrosis, atypical mitotic figures, and hemorrhage were also seen. Six cases contained a myxoid matrix (Figure 1c). Lobulated or multinodular architecture, granulomatous appearance, metaplastic elements, abundant hyalinized or chondromyxoid matrix, or paracholdoma-like vacuolated cytoplasm were not evident.

Conventional-type tumors. Rhabdoid cells were arranged in a sheet (a, 2-year-old female, back) or alveolar (b, 2-year-old male, kidney pattern). A small nested pattern with focal myxoid matrix was also observed (c, 1-month-old male, thigh).
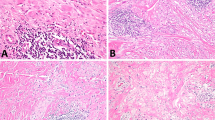
The second type, into which 10 cases were classified (4 of the 14 cases of malignant rhabdoid tumor of the kidney; 2 of the 19 cases of extrarenal malignant rhabdoid tumor; 2 of the 11 cases of atypical teratoid/rhabdoid tumor; and 2 of the 8 cases of undifferentiated/unclassified sarcoma) resembled atypical teratoid/rhabdoid tumor with mesenchymal or epithelial features. This atypical teratoid/rhabdoid type showed a proliferation of epithelioid tumor cells having large, vesicular, and oval-to-polygonal nuclei, prominent nucleoli, ;and scant-to-abundant eosinophilic cytoplasm, arranged in sheets or in solid trabecular or solid alveolar patterns, accompanied by rhabdoid cells. The majority of tumors of this type showed tumor necrosis, atypical mitotic figures, and hemorrhage. Spindle-shaped tumor cell components were also recognized in 9 of the 10 atypical teratoid/rhabdoid-type cases (Figure 2a). Two cases of undifferentiated/unclassified sarcoma showed epithelial-like components arranged in rosette, pseudoglandular, or cord patterns (Figure 2b). One case of undifferentiated/unclassified sarcoma showed squamoid cell nests (Figure 2c). Two cases contained a myxoid matrix. Lobulated or multinodular architecture, granulomatous appearance, and parachordoma-like vacuolated cytoplasm were not evident.

Atypical teratoid/rhabdoid-type tumors. Spindle-shaped cells were arranged in a fascicular pattern (a, 1-year-old female, kidney). Epithelioid cells were arranged in tubular or glandular structures (b, newborn male, liver). Squamoid cells having abundant clear-to-eosinophilic cytoplasms and sharp cell boundaries arranged in a nested pattern were observed (c, newborn male, temple).
The third type, into which six cases were classified (six of the eight undifferentiated/unclassified sarcoma cases), were malignant lymphoma-like small round cell tumors without rhabdoid cells. This small cell type showed a proliferation of uniform small round tumor cells having hyperchromatic nuclei, prominent nucleoli, and scant cytoplasms arranged in sheets (Figure 3). The majority of tumors of this type showed tumor necrosis, atypical mitotic figures, and hemorrhage. Rhabdoid cells, lobulated or multinodular architecture, granulomatous appearance, metaplastic elements, and abundant hyalinized or chondromyxoid matrix were not evident.

Small cell-type tumors. Uniform small rounded cells with scant cytoplasm were arranged in a sheet pattern (4-year-old male, retroperitoneum).
Immunohistochemical Findings
The results of the immunohistochemical analysis are summarized in Tables 4a and 4b. All of the available tumor specimens were positive for vimentin and epithelial marker (EMA, CAM5.2, or AE1/AE3) (Figures 4a–c). In particular, epithelial-like components of atypical teratoid/rhabdoid type were diffusely and strongly positive for these epithelial markers, but not in the globular pattern of staining typical of malignant rhabdoid tumor. Positivity for α-SMA was observed in 63% of conventional-type and 67% of atypical teratoid/rhabdoid-type tumors, but in only 25% of small cell-type tumors. Only two cases were positive for desmin. Three of the 30 (10%) reclassified cases were positive for ERG, but positivity for SALL4 and GPC3 was seen in only 19 of the 31 (58%) and 22 of the 37 (59%) reclassified cases, respectively. Twelve of the 38 (32%) reclassified cases showed CD34 immunoexpression (Figure 4d).

The cytoplasms of tumor cells in the conventional-type category were positive for vimentin (a, 2-year-old male, kidney), CAM5.2 (c, 4-month-old male, liver) and CD34 (d, 5-month-old female, kidney). Focal vimentin immunoreactivity is evident in the nuclear periphery of small cell-type tumor cells (b, 4-year-old male, retroperitoneum).
MicroRNA Array Analysis
Five of the cases of malignant rhabdoid tumor and one case of pediatric undifferentiated/unclassified small round cell sarcoma (Table 1, case 5) were reclassified as conventional- and small cell-type tumors, respectively.
An unsupervised hierarchical clustering based on the relative expression of microRNAs with valid duplicate spots showed distinctive expression profiles for tumor samples and non-tumorous skeletal muscle samples (Pearson's correlation coefficient, 0.795641). However, the microRNA profiles in the MRTs (reclassified conventional-type) and pediatric undifferentiated/unclassified small round cell sarcoma (reclassified small cell-type) were intermingled (Pearson's correlation coefficient, 0.888300 or 0.891388) (Figure 5).

Global microRNA expression and unsupervised hierarchical clustering of malignant rhabdoid tumor (MRT), undifferentiated/unclassified small cell sarcoma (US), and non-tumorous skeletal muscle (NSM). US and MRT have very close microRNA profiles (Pearson’s correlation coefficient, 0.888300 or 0.891388).
Follow-up
Follow-up data were available in 37 malignant rhabdoid tumor cases (kidney, 11 cases; soft tissue and liver, 15 cases; central nervous system, 11 cases) and 7 undifferentiated/unclassified sarcoma cases with a mean follow-up duration of 26 months (range 1–204 months). The overall survival rate was not significantly different between initially diagnosed atypical teratoid/rhabdoid tumor of the central nervous system and malignant rhabdoid tumor of the extra-central nervous system (P=0.16) (Figure 6a). There were also no statistically significant differences in any of the reclassified combinations (conventional type vs atypical teratoid/rhabdoid type, P=0.73; conventional type vs small cell type, P=0.58; atypical teratoid/rhabdoid type vs small cell type, P=0.74) (Figure 6b).

Overall survival curves of patients with the existing classifications (a, malignant rhabdoid tumor vs atypical teratoid/rhabdoid tumor) and patients assigned to our three reclassified groups (b, conventional-, atypical teratoid/rhabdoid- and small cell-type tumors). There were no statistically significant differences among any of the combinations.
Discussion
Malignant rhabdoid tumor has been shown to occur in a wide variety of extrarenal sites and in the central nervous system.15 The first example affecting the central nervous system was simply called rhabdoid tumor.5 These tumors were named atypical teratoid/rhabdoid tumor when they occurred in the central nervous system to call attention to the disparate combination of rhabdoid, primitive neuroepithelial, epithelial, and mesenchymal components.5, 16, 17 However, in our present study we found six cases of extra-central nervous system malignant rhabdoid tumor with atypical teratoid/rhabdoid tumor-like features and nine cases of central nervous system atypical teratoid/rhabdoid tumor with conventional malignant rhabdoid tumor-like features. In addition, two cases of extra-central nervous system undifferentiated/unclassified sarcoma with massively epithelial and mesenchymal components were recognized. Although epithelial and mesenchymal features such as glandular pattern or sarcomatoid spindle-shaped tumor cells are not included under the category of extrarenal rhabdoid tumor in the World Health Organization classification of bone and soft-tissue tumors, this current study shows that these features can actually be seen in extrarenal malignant rhabdoid tumor.12
As for the immunohistochemical profile and prognosis, there were no significant differences between the two existing classifications: rhabdoid tumor of the extra-central nervous system (malignant rhabdoid tumor) and rhabdoid tumor of the central nervous system (atypical teratoid/rhabdoid tumor). In our reclassified criteria as well, no statistically significant differences in prognosis were found. However, immunoreactivity for cytokeratins (CAM5.2 and AE1/AE3), smooth muscle marker (α-SMA) or neural marker (S-100 protein) was more frequent in the conventional or atypical teratoid/rhabdoid type than in the small cell type. The small cell type may be a more poorly differentiated form compared with the conventional type and atypical teratoid/rhabdoid type, and our present data suggested the present criteria based on histological features. Therefore, it seemed desirable that rhabdoid tumor be reclassified according to histological characteristics rather than the anatomic sites of occurrence.
With respect to the differential diagnosis, the histological features of malignant rhabdoid tumor particularly resemble those of epithelioid sarcoma, especially proximal-type epithelioid sarcoma. At present, immunoreactivity for CD34 is generally accepted as a differential marker; approximately half of all epithelioid sarcoma cases are positive for CD34, whereas malignant rhabdoid tumor cases scarcely reveal immunoreactivity for this marker.18, 19, 20, 21, 22, 23 In the present study, CD34 immunoreactivity was found in 12 of the 38 rhabdoid tumor cases (31%), and the previously reported immunoreactive ratio was 60%.23 Therefore, CD34 may not be useful as a differential diagnostic marker. On the other hand, the immunoreactive ratios for ERG, SALL4, or GPC3 in the current reclassified rhabdoid tumor cases, including our previously reported cases, were 10%, 61%, and 59%, respectively. In the previously reported proximal-type epithelioid sarcoma cases, ERG, SALL4, or GPC3 immunoexpression was found in 25%, 0–25%, or 5% of cases, respectively.22, 23 Combined evaluations of the immunoreactivities for ERG, SALL4, and GPC3 may thus be a useful diagnostic tool to distinguish rhabdoid tumor from proximal-type epithelioid sarcoma.
Undifferentiated/unclassified sarcoma is defined as a sarcoma with no identifiable line of differentiation when analyzed by presently available technology.11 These tumors show various histologic features and the diagnosis is one of exclusion after application of close morphological observation and judicious use of ancillary techniques. In the present study, eight cases of pediatric undifferentiated/unclassified sarcoma were found to have complete loss of SMARCB1/INI1 expression, and could be classified into two categories, atypical teratoid/rhabdoid type and small cell type, based on their histological features.
We performed microRNA array analysis to confirm the correspondence between the small cell-type tumors and malignant rhabdoid tumor. MicroRNAs exert multiple biological functions by negatively regulating the expression of their target genes involved in development, differentiation, apoptosis, and cell proliferation, and it is suggested that knowledge of microRNA expression profiles in cancer may have substantial value for diagnostic and prognostic determinations.14, 24, 25, 26 In our current study, the microRNA profiles in the malignant rhabdoid tumor group (histologically conventional type) and undifferentiated/unclassified small round cell tumors with complete loss of SMARCB1/INI1 protein expression were intermingled. Further, the overall survival of the patients with small cell-type tumors was in the same range as the survival of patients classified into the conventional-type and atypical teratoid/rhabdoid-type groups. Therefore, it was suggested that the small cell-type classification may be considered a morphological variant of the definition of malignant rhabdoid tumor. However, some malignant rhabdoid tumor cases demonstrate predominant proliferation of undifferentiated small round cells with only a small number of typical rhabdoid cells.12 In particular, limited biopsies may fail to sample rhabdoid cells. Thus, in the case of diagnosing tumors of small cell type, detailed histological and immunohistochemical examinations are necessary.
In the previous report, there were five cases of pediatric small round cell tumor with loss of SMARCB1/INI1 protein expression resembling our cases of small cell type.7 One of the five cases did not have rhabdoid cells with intracytoplasmic inclusions at diagnosis, but did display rhabdoid cells at definitive surgery following chemotherapy.7 Therefore, it seems that these cases may be on the same spectrum as malignant rhabdoid tumor, although the outcomes were certainly more favorable than those for malignant rhabdoid tumor: two of these patients are alive and well after more than 15 years and the remaining three cases are alive and well but with <2 years of follow-up.7 In our present study, one patient with small cell-type tumor has been alive with no evidence of disease for 80 months, and another case died of causes related to treatment after 15 months, carrying no residual tumor at the time of death. However, the remaining three patients with small cell-type tumor died of disease within 8 months. There were no statistically significant differences in prognosis between the small cell-type group and the other groups (conventional-type and atypical teratoid/rhabdoid-type).
In summary, we analyzed clinicopathological features in cases of malignant rhabdoid tumor, atypical teratoid/rhabdoid tumor, and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 expression, and reclassified them into three groups according to their histological features: conventional-, atypical teratoid/rhabdoid-, and small cell-type tumors. Malignant rhabdoid tumor (rhabdoid tumors in the extra-central nervous system) with teratoid features and atypical teratoid/rhabdoid tumor (rhabdoid tumor in the central nervous system) without teratoid features cases were identified. We were able to demonstrate a very close lineage between the cases of pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 protein expression and the two rhabdoid tumor (malignant rhabdoid tumor and atypical teratoid/rhabdoid tumor) groups. We suggest that reclassification of malignant rhabdoid tumor into three groups according to histologic features rather than the traditional classification by sites of organ would be favorable for their histopathological diagnosis.
References
Wilson BG, Roberts CW . SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011;11:481–492.
Hollmann TJ, Hornick JL . INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol 2011;35:e47–e63.
Guidi CJ, Sands AT, Zambrowicz BP et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol 2001;21:3598–3603.
Beckwith JB, Palmer NF . Histopathology and prognosis of Wilms tumors: results from the First National Wilms' Tumor Study. Cancer 1978;41:1937–1948.
Judkins AR, Eberhart CG, Wesseling P . Atypical teratoid/rhabdoid tumor. In: Louis DN, Ohgaki H, Wiestler OD et al (eds). World Health Organization Classification of Tumours of Central Nervous System. IARC Press: Lyon, France, 2007, pp 147–149.
Fletcher CDM, Antonescu CR, Heim S et al. Myoepithelioma/myoepithelial carcinoma/mixed tumour. In: Fletcher CDM, Bridge JA, Hogendoorn P et al (eds). World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press: Lyon, France, 2013, pp 153–154.
Kreiger PA, Judkins AR, Russo PA et al. Loss of INI1 expression defines a unique subset of pediatric undifferentiated soft tissue sarcomas. Mod Pathol 2009;22:142–150.
Hasselblatt M, Gesk S, Oyen F et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 2011;35:933–935.
Hachitanda Y, Tsuneyoshi M, Enjoji M et al. Congenital primitive neuroectodermal tumor with epithelial and glial differentiation. An ultrastructural and immunohistochemical study. Arch Pathol Lab Med 1990;114:101–105.
Ohyama M, Ijiri R, Tanaka Y et al. Congenital primitive epithelial tumor of the liver showing focal rhabdoid features, placental involvement, and clinical features mimicking multifocal hemangioma or stage 4 S neuroblastoma. Hum Pathol 2000;31:259–263.
Fletcher CDM, Chibon F, Mertens F et al. Undifferentiated/unclassified sarcomas. In: Fletcher CDM, Bridge JA, Hogendoorn P et al (eds). World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press: Lyon, France, 2013, pp 236–238.
Oda Y, Biegel JA . Extrarenal rhabdoid tumour. In: Fletcher CDM, Bridge JA, Hogendoorn P (eds). World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press: Lyon, France, 2013, pp 228–229.
Yamamoto H, Kohashi K, Fujita A et al. Fascin-1 overexpression and miR-133b downregulation in the progression of gastrointestinal stromal tumor. Mod Pathol 2013;26:563–571.
Kohashi K, Yamamoto H, Kumagai R et al. Differential microRNA expression profiles between malignant rhabdoid tumor and epithelioid sarcoma: miR193a-5p is suggested to downregulate SMARCB1 mRNA expression. Mod Pathol 2014;27:832–839.
Oda Y, Tsuneyoshi M . Extrarenal rhabdoid tumors of soft tissue: clinicopathological and molecular genetic review and distinction from other soft-tissue sarcomas with rhabdoid features. Pathol Int 2006;56:287–295.
Rorke LB, Packer R, Biegel J . Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol 1995;24:21–28.
Rorke LB, Packer RJ, Biegel JA . Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 1996;85:56–65.
Oda Y, Dal Cin P, Laskin WB . Epithelioid sarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn P et al (eds). World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press: Lyon, France, 2013, pp 216–218.
Izumi T, Oda Y, Hasegawa T et al. Prognostic significance of dysadherin expression in epithelioid sarcoma and its diagnostic utility in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Mod Pathol 2006;19:820–831.
Kohashi K, Izumi T, Oda Y et al. Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: a useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol 2009;40:349–355.
Kohashi K, Nakatsura T, Kinoshita Y et al. Glypican 3 expression in tumors with loss of SMARCB1/INI1 protein expression. Hum Pathol 2013;44:526–533.
Kohashi K, Yamada Y, Hotokebuchi Y et al. ERG and SALL4 expressions in SMARCB1/INI1-deficient tumors: a useful tool for distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol 2015;46:225–230.
Yoshida A, Asano N, Kawai A et al. Differential SALL4 immunoexpression in malignant rhabdoid tumours and epithelioid sarcomas. Histopathology 2015;66:252–261.
Calin GA, Croce CM . MicroRNA signatures in human cancers. Nat Rev Cancer 2006;6:857–866.
Hisaoka M, Matsuyama A, Nagao Y et al. Identification of altered MicroRNA expression patterns in synovial sarcoma. Genes Chromosomes Cancer 2011;50:137–145.
Subramanian S, Lui WO, Lee CH et al. MicroRNA expression signature of human sarcomas. Oncogene 2008;27:2015–2026.
Acknowledgements
This study was supported by JSPS KAKENHI Grant Numbers 25293088 and 26460435, and by a grant for Clinical Research from the Ministry of Health Labour and Welfare, Tokyo, Japan. The English used in this manuscript was revised by KN International (http://www.kninter.com/).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Presented in part at the 102nd annual meeting of the United States and Canadian academy of Pathology in Baltimore, Maryland, 2–8 March 2013.
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Kohashi, K., Tanaka, Y., Kishimoto, H. et al. Reclassification of rhabdoid tumor and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 protein expression: three subtypes of rhabdoid tumor according to their histological features. Mod Pathol 29, 1232–1242 (2016). https://doi.org/10.1038/modpathol.2016.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.106
This article is cited by
-
Establishment and characterization of NCC-MRT1-C1: a novel cell line of malignant rhabdoid tumor
Human Cell (2022)
-
SWI/SNF-deficient undifferentiated/rhabdoid carcinoma of the gallbladder carrying a POLE mutation in a 30-year-old woman: a case report
Diagnostic Pathology (2021)
-
Soft Tissue Special Issue: Skeletal Muscle Tumors: A Clinicopathological Review
Head and Neck Pathology (2020)
-
No compelling need to reclassify
Modern Pathology (2017)
