
Abstract
Asthma is a complex disease of the lungs, which is characterized by airway inflammation and airway hyperresponsiveness (AHR). Alveolar macrophages (AMs), one of the prominent immune system cells found in the airways, have been implicated in the development and progression of asthma. AMs constitute a unique subset of pulmonary macrophages, which serve as a first line of defense against foreign invaders to the lung tissue. In addition, based on human and animal studies, they have also been found to regulate pro- and anti-inflammatory responses in the airways, suggesting that these cells have a critical role in asthma. In this review, our focus is to evaluate the relevance of AMs in the context of asthma, and the underlying mechanisms that regulate their functions.
Similar content being viewed by others

Introduction
Asthma is a chronic inflammatory disease characterized by bronchial hyperresponsiveness, airway remodeling, and increased influx of inflammatory cells in the lungs along with a prominent Th2 cytokine signature.1
Among inflammatory cells, macrophages are the most abundant leukocytes found in alveoli, distal airspaces, and conducting airways,2, 3, 4 suggesting that they have an important role in providing protection against foreign pathogens at the interface of the external environment and the alveolar tissue. Existing literature has established a substantial link between lung macrophages and airway remodeling5, 6 and eosinophilic inflammation6 in asthma. However, it is not yet clear whether these cells have an inflammatory or immunosuppressive role.
Lung Macrophages and Asthma
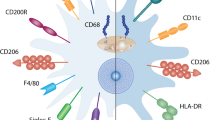
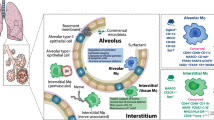
Mainly, three classes of macrophages are identified in the lung tissue: bronchial macrophages (BMs), alveolar macrophages (AMs), and interstitial macrophages (IMs). BMs are isolated from induced sputum,7, 8 while AMs are found in alveoli lumen, presiding a nonspecific innate defense mechanism. IMs inhabit the interstitium where they interact with interstitial lymphocytes and help in mounting a specific immune response.9, 10, 11 Generally IMs are considered as antigen-presenting macrophages,12 but when AMs are primed with allergen ex-vivo, they can also help in expanding allergen-specific T-cell response in human and guinea pigs.13, 14, 15 Studies on rodents, however, do not go along with the concept of AMs as antigen-presenting macrophages.16 Given that BMs, AMs, and IMs occupy different compartments in lungs, it would be interesting to understand whether they collaborate to mediate their functions. The aim of this article is to provide a circumspect review of the available data on AMs specifically and to discuss their role in the development of pulmonary pathology in asthma.
AMs arise from circulating blood monocytes, which colonize the tissues under inflammatory and noninflammatory states. AM-derived monocytes chemoattractant protein-1 has a significant role in the recruitment of monocytes to the inflamed tissue.17, 18 AMs are also shown to originate from IMs, which serve as obligate intermediate between blood monocytes and AMs.19, 20 Local proliferation sustained by colony-stimulating factor is also suggested as an important source of their expansion in human and murine airways.21, 22 There is one more type of lung macrophages described in humans, cats, dogs, and sheep, known as intravascular macrophages. Intravascular macrophages are located on the inner side of capillaries, suggested to fight against pathogens that are entering into the lungs through blood. These cells are, however, not found in rodents.23, 24, 25 As their function is comparable to that of AMs, it would be interesting to determine whether they serve as an intermediate stage between blood monocytes and AMs, or they actually exist as a discrete population.
Macrophages, in general (including AMs and IMs), are characterized based upon their role and cytokine profile. Two such subpopulations are currently known: M1 and M2 subsets.26 M1 macrophages are known to drive inflammation in response to intracellular pathogens,27, 28, 29 while M2 macrophages are involved in phagocytosis of foreign pathogens and apoptotic cells.30 Previously, interferon-γ and lipopolysaccharide were thought to polarize macrophage differentiation to M1 while Th2 cytokines interleukin (IL)-4 and IL-13 to M2 type of macrophages.27, 28, 29, 30 Interferon-γ in combination with lipopolysaccharide or IL-27 exaggerates airway hyperresponsiveness (AHR) and non-eosinophilic inflammation.31 As these conditions also induce M1 polarization, this effect is likely to be mediated partially through M1 cells.
Th2 cytokines skews macrophage differentiation to M2 type. M2 cells secrete FIZZ1 (found in inflammatory zone-1) and Ym-1 (a chitinase-like molecule) upon Aspergillus fumigatus challenge in asthmatic mice.29, 32 FIZZ1 (a resistin-like molecule-α) and Ym-1,33 induced by Th2 cytokines, are overexpressed in asthma.19 The production of these profibrotic and inflammatory factors is infact a defense mechanism to maintain pulmonary homeostasis in asthma; however, their excessive production results in aggravated lung injury.19
Recent developments have, however, suggested plasticity in macrophage phenotypes as reviewed in ref. 34. It is due to the overlapping of phenotypes and conversion of one phenotype to another depending upon the inflammatory response; it is not clear which subpopulation is most involved in asthma pathogenesis or resolution. There are studies that showed promotion of both M1 and M2 cell types irrespective of the cytokine mileau. For example, IL-33 promotes a Th2-type response35, 36 while activating both subtypes of macrophages and contributing to allergic inflammation.37, 38, 39 In a related study, it was found that IL-4 could also induce the production of IL-6, IL-12p70, and tumor necrosis factor by M1 macrophages.39 Collectively, more studies are needed to clarify the role of macrophage phenotypes plasticity in asthma.
Anti-Inflammatory Role of AMs
Anatomic location of AMs and their functions suggest that they are capable of maintaining homeostasis in the airways. On one hand, they fight against pathogens by activating multiple immunological pathways and serve as first line of defense, while on the other hand, they manifest an anti-inflammatory response to protect excessive tissue damage. Such anti-inflammatory response is observed in several asthmatic studies too. Depletion of macrophages and subsequent adoptive transfer of these cells from genetically or phenotypically dissimilar donors have been widely used as a principle strategy to study their functions in vivo.40, 41, 42 A significant percentage of AMs can be depleted by intratracheal or intranasal administration of the drug, clodronate, encapsulated in liposome.40, 41 Clodronate thus administered is phagocytosed by AMs, which degrade the drug using its lysosomal machinery. The degraded product then disrupts the mitochondrial inner membrane integrity and induces apoptosis in AMs.43 Here, depletion of other phagocytic cells such as dendritic cells and neutrophils is a major concern; however, it is less likely due to their inability to degrade clodronate pertaining to different phagocytic mechanisms employed by them.44 By adoptive transfer experiments, it was found that allergen induced AHR and inflammation is ameliorated when AMs from unsensitized mice are transferred to macrophage-depleted sensitized mice.42 It was apparent that AMs from unsensitized mice, which are not antigen-primed and have better regeneration capacity,45, 46 provided protection against asthmatic symptoms. Earlier studies also indicated the role of asthma-associated immune dysfunctions in affecting early precursors of AMs.42 A study by Vissers et al.47 further demonstrated that intravenously administered allergen-loaded macrophages accumulate in the spleen and induce an indirect but long-term allergen-specific immunosuppressive effect through regulatory T cells. Although these macrophages are not AMs and do not cumulate in the lungs, the study suggests that allergen priming of macrophages can also manifest a protective function. Their finding is further supported by a report by Bang et al.,42 which showed that AM depletion worsened AHR and inflammation in asthmatic mice. Brown Norway rats also showed similar response on AM depletion.48 AMs are rather found to control nonspecific inflammatory response.48 Altogether, it seems that AMs have a protective role; however, AMs from asthmatics may not be as efficient as those from healthy non-asthmatics in contributing to an anti-inflammatory response. Therefore, transfer of unsensitized AMs to AM-depleted asthmatic mice is found to provide protection as they allow apposite phagocytosis of apoptotic cells so that super-proinflamatory functions of these dying cells and other antigen-presenting cells can be suppressed.49, 50 The regulatory control of AMs on T cells is mediated by suppressing their proliferation through a nitric oxide synthase-dependent pathway.51, 52 Suppression of DC-induced T-cell activation by AMs has also been reported.50 However, this suppressive effect on lymphocytes can be reversed by granulocyte–monocyte colony-stimulating factor, produced locally during acute inflammatory reaction.53
Similar observations were made in human asthmatic studies. Suppression of T-cell proliferation by AMs was proposed to be mediated through cell–cell interaction.51 Further, AMs from non-asthmatic subjects express lesser co-stimulatory molecules (CD80 and CD86) compared with AMs from asthmatics, and thus contain lesser potential to induce the production of IL-4 and IL-5 by T-cell lines.54, 55 Moreover, AMs from asthmatic subjects produce greater IL-10 than healthy counterparts, which could be an additional mechanism to suppress the inflammatory response. IL-10 production by AMs in asthma can be further increased by corticosteroid treatment.56 Enhanced production of IL-12 by AMs from asthmatics than their healthy counterparts was also proposed to neutralize Th2 response.56 However, this report was contradicted by a succeeding reminiscent study,57 therefore status of AMs in regulating the expression of IL-12 is uncertain. AMs, which constitutively produce nitric oxide, do so at greater extent in asthmatic state. Enhanced nitric oxide thus produced inhibits the production of inflammatory cytokines from AMs.58 Current evidence (as briefed in Table 1) illustrates the immunosuppressive effects of macrophages to confer resistance to asthmatic manifestations and suggests that this resistance is transferable.
Pro-Inflammatory Role of AMs
Current literature has also indicated an essential role of AMs in the development of allergic airway diseases such as asthma. Human and animal studies have provided crucial information on how activation of AMs drives Th2 inflammation. Besides dysfunctional phenotype of such cells in asthma, their role in exacerbating IgE-dependent inflammation has also been observed.
In a study by Careau et al.,49 it was observed that when allergen-primed AMs are transferred to AM-depleted non-asthmatic healthy mice, they do not induce AHR and inflammation.49 Contrary to this, Moon et al.6 demonstrated that AMs from sensitized mice are capable of inducing eosinophilic inflammation upon transfer to unsensitized mice. The disparity in the observations may be owing to differences in the experimental strategies employed in these studies. For example, in the later report, nonsensitized non-asthmatic mice are challenged with ovalbumin (OVA) after adoptive transfer of sensitized AMs. As the origin of AMs in this study was not studied, it is possible that eosinophilic inflammation, which was observed in the recipient animals, resulted from immune activation owing to OVA challenge and not entirely due to the adoptively transferred sensitized AMs. This study, however, draws attention to another side of the coin: a proinflammatory role of AMs. Sensitization with allergen increases the percentage of CCR3-expressing CD11b+ CD11cintermediate macrophages, which are otherwise CD11b intermediate CD11c+ AMs in normal animals.6 Allergen exposure renders these AMs more efficient in stimulating allergen-specific T cells, thereby resulting in an elaboration of the Th2 cytokines (IL-4, IL-5, and IL-13) in bronchoalveolar lavage fluid. OVA sensitization followed by challenge results in IL-17 overexpression by CD11b+F4/80+ AMs, which mediate OVA-induced AHR and allergic inflammation in these animals.59 Infact, OVA challenge induces the release of mediators by mast cells, which then induce IL-17 production by AMs.59
AMs facilitate the recruitment of activated neutrophils to the alveolar space.60 Activated neutrophils have a short inflammatory life span as they are programed to undergo constitutive apoptosis within few hours of activation.61 Apoptotic neutrophils are recognized and phagocytosed by AMs.62 This process itself helps resolve pulmonary inflammation, first by preventing the leakage of toxic neutrophilic intracellular contents, and second by inducing the production of IL-10. However, paralysed phagocytic capacity of AMs is observed in asthmatic subjects, which further amplifies the inflammation by not efficiently clearing the dying cells. Downregulation of CD16 and CD64 was observed in BMs isolated from asthmatic individuals as compared with healthy subjects. CD16 and CD64 mediate the phagocytic activity of BMs, their poor expression results in compromised phagocytic activity of BMs.8, 63 However, whether a similar phenomenon is occurring in AMs is not known.
Role of IgE and its low- and high-affinity receptors have also been observed in mediating asthmatic symptoms by AMs. IgE-induced CD23-dependent production of proinflamatory and anti-inflammatory cytokines was observed (Table 1). The effect of IgE was tested on control and asthmatic subjects, and it was found that AMs from asthmatic subjects showed greater production of proinflamatory cytokines than anti-inflammatory cytokines.64 Stimulation with IgE–anti IgE complex induces the production of leukotriene B4, prostaglandin F2cx, thromboxane B2, and the lysosomal hydrolase N-acetyl-fi-d-glucosaminidase.65 Other products from arachidonic acid pathway, leukotriene C4 and thromboxane A2 are also induced by AMs, potentially modulate the bronchial smooth muscle tone.66, 67 IgE–anti IgE complex induces the production of tumor necrosis factor by AMs, which is further amplified by pretreatment with interferon-γ.68 Collectively, there are indications of direct and indirect role of AMs, contributing to the inflammation during asthmatic manifestations.
Concluding Remarks
Current evidence suggests that AMs have a wide range of immunoregulatory functions in asthma beyond that of a barrier to pathogens intruding into the alveolar tissue. The studies discussed in this review assert that macrophages are not only associated with the maintenance of pulmonary homeostasis by performing anti-inflammatory functions, but also likely an integral part of the mechanisms perpetuating inflammation and tissue injury associated with asthma. However, further research focusing on the modulatory relationship between AMs and asthma in a unified model of asthma, irrespective of the genetic differences of experimental animals, is warranted. Moreover, studies to determine universal cell surface markers of AMs would improve our understanding of the type of AMs and help to unravel their role in the pathogenesis of asthma.
References
Robinson, D.S. The role of the T cell in asthma. J. Allergy. Clin. Immunol. 126, 1081–1091; quiz 1092-1083 (2010).
Arjomandi, M. et al. Repeated exposure to ozone increases alveolar macrophage recruitment into asthmatic airways. Am. J. Respir. Crit. Care. Med. 172, 427–432 (2005).
Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 (2003).
Leung, T.F., Wong, G.W., Ko, F.W., Lam, C.W. & Fok, T.F. Increased macrophage-derived chemokine in exhaled breath condensate and plasma from children with asthma. Clin. Exp. Allergy. 34, 786–791 (2004).
Mautino, G. et al. Increased expression of tissue inhibitor of metalloproteinase-1 and loss of correlation with matrix metalloproteinase-9 by macrophages in asthma. Lab. Invest. 79, 39–47 (1999).
Moon, K.A. et al. Allergen-induced CD11b+ CD11c(int) CCR3+ macrophages in the lung promote eosinophilic airway inflammation in a mouse asthma model. Int. Immunol. 19, 1371–1381 (2007).
Pizzichini, E., Pizzichini, M.M., Leigh, R., Djukanovic, R. & Sterk, P.J. Safety of sputum induction. Eur. Respir. J. Suppl. 37, 9s–18s (2002).
Moniuszko, M., Bodzenta-Lukaszyk, A., Kowal, K. & Dabrowska, M. Bronchial macrophages in asthmatics reveal decreased CD16 expression and substantial levels of receptors for IL-10, but not IL-4 and IL-7. Folia. Histochem. Cytobiol. 45, 181–189 (2007).
Franke-Ullmann, G. et al. Characterization of murine lung interstitial macrophages in comparison with alveolar macrophages in vitro. J. Immunol. 157, 3097–3104 (1996).
Prokhorova, S., Lavnikova, N. & Laskin, D.L. Functional characterization of interstitial macrophages and subpopulations of alveolar macrophages from rat lung. J. Leukoc. Biol. 55, 141–146 (1994).
Fathi, M. et al. Functional and morphological differences between human alveolar and interstitial macrophages. Exp. Mol. Pathol. 70, 77–82 (2001).
Gong, J.L., McCarthy, K.M., Rogers, R.A. & Schneeberger, E.E. Interstitial lung macrophages interact with dendritic cells to present antigenic peptides derived from particulate antigens to T cells. Immunology 81, 343–351 (1994).
Venet, A., Hance, A.J., Saltini, C., Robinson, B.W. & Crystal, R.G. Enhanced alveolar macrophage-mediated antigen-induced T-lymphocyte proliferation in sarcoidosis. J. Clin. Invest. 75, 293–301 (1985).
Lyons, C.R. & Lipscomb, M.F. Alveolar macrophages in pulmonary immune responses. I. Role in the initiation of primary immune responses and in the selective recruitment of T lymphocytes to the lung. J. Immunol. 130, 1113–1119 (1983).
Toews, G.B. et al. The accessory cell function of human alveolar macrophages in specific T cell proliferation. J. Immunol. 132, 181–186 (1984).
Holt, P.G. & Batty, J.E. Alveolar macrophages. V. Comparative studies on the antigen presentation activity of guinea-pig and rat alveolar macrophages. Immunology 41, 361–366 (1980).
Brieland, J.K. et al. Effect of acute inflammatory lung injury on the expression of monocyte chemoattractant protein-1 (MCP-1) in rat pulmonary alveolar macrophages. Am. J. Respir. Cell. Mol. Biol. 7, 134–139 (1992).
Jiang, Y., Beller, D.I., Frendl, G. & Graves, D.T. Monocyte chemoattractant protein-1 regulates adhesion molecule expression and cytokine production in human monocytes. J. Immunol. 148, 2423–2428 (1992).
Moreira, A.P. & Hogaboam, C.M. Macrophages in allergic asthma: fine-tuning their pro- and anti-inflammatory actions for disease resolution. J. Interferon. Cytokine. Res. 31, 485–491 (2011).
Landsman, L. & Jung, S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J. Immunol. 179, 3488–3494 (2007).
Lin, H.S., Lokeshwar, B.L. & Hsu, S. Both granulocyte-macrophage CSF and macrophage CSF control the proliferation and survival of the same subset of alveolar macrophages. J. Immunol. 142, 515–519 (1989).
Nakata, K. et al. Granulocyte-macrophage colony-stimulating factor promotes the proliferation of human alveolar macrophages in vitro. J. Immunol. 147, 1266–1272 (1991).
Warner, A.E., Barry, B.E. & Brain, J.D. Pulmonary intravascular macrophages in sheep. Morphology and function of a novel constituent of the mononuclear phagocyte system. Lab. Invest. 55, 276–288 (1986).
Warner, A.E., Molina, R.M. & Brain, J.D. Uptake of bloodborne bacteria by pulmonary intravascular macrophages and consequent inflammatory responses in sheep. Am. Rev. Respir. Dis. 136, 683–690 (1987).
Dehring, D.J. & Wismar, B.L. Intravascular macrophages in pulmonary capillaries of humans. Am. Rev. Respir. Dis. 139, 1027–1029 (1989).
Mantovani, A., Garlanda, C. & Locati, M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler. Thromb. Vasc. Biol. 29, 1419–1423 (2009).
Cassol, E., Cassetta, L., Rizzi, C., Alfano, M. & Poli, G. M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J. Immunol. 182, 6237–6246 (2009).
Kurata, S., Matsumoto, M., Tsuji, Y. & Nakajima, H. Lipopolysaccharide activates transcription of the heme oxygenase gene in mouse M1 cells through oxidative activation of nuclear factor kappa B. Eur. J. Biochem. 239, 566–571 (1996).
Moreira, A.P. et al. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J. Allergy. Clin. Immunol. 126, 712–721, e717.
Mills, C.D., Kincaid, K., Alt, J.M., Heilman, M.J. & Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 164, 6166–6173 (2000).
Li, J.J. et al. IL-27/IFN-gamma induce MyD88-dependent steroid-resistant airway hyperresponsiveness by inhibiting glucocorticoid signaling in macrophages. J. Immunol. 185, 4401–4409 (2010).
Zhang, L. et al. Oxidative stress and asthma: proteome analysis of chitinase-like proteins and FIZZ1 in lung tissue and bronchoalveolar lavage fluid. J. Proteome. Res. 8, 1631–1638 (2009).
Chupp, G.L. et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. N. Engl. J. Med. 357, 2016–2027 (2007).
Mosser, D.M. & Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008).
Schmitz, J. et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490 (2005).
Kearley, J., Buckland, K.F., Mathie, S.A. & Lloyd, C.M. Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2-IL-33 pathway. Am. J. Respir. Crit. Care. Med. 179, 772–781 (2009).
Kurowska-Stolarska, M. et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 183, 6469–6477 (2009).
Joshi, A.D. et al. Interleukin-33 contributes to both M1 and M2 chemokine marker expression in human macrophages. BMC Immunol. 11, 52 (2010).
Varin, A., Mukhopadhyay, S., Herbein, G. & Gordon, S. Alternative activation of macrophages by IL-4 impairs phagocytosis of pathogens but potentiates microbial-induced signalling and cytokine secretion. Blood 115, 353–362 (2010).
Thepen, T., Van Rooijen, N. & Kraal, G. Alveolar macrophage elimination in vivo is associated with an increase in pulmonary immune response in mice. J. Exp. Med. 170, 499–509 (1989).
Wu, M. et al. Genetically engineered macrophages expressing IFN-gamma restore alveolar immune function in scid mice. Proc. Natl Acad. Sci. USA 98, 14589–14594 (2001).
Bang, B.R. et al. Alveolar macrophages modulate allergic inflammation in a murine model of asthma. Exp. Mol. Med. 43, 275–280.
Lehenkari, P.P. et al. Further insight into mechanism of action of clodronate: inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol. Pharmacol. 61, 1255–1262 (2002).
Savina, A. & Amigorena, S. Phagocytosis and antigen presentation in dendritic cells. Immunol. Rev. 219, 143–156 (2007).
Fitzpatrick, A.M., Holguin, F., Teague, W.G. & Brown, L.A. Alveolar macrophage phagocytosis is impaired in children with poorly controlled asthma. J. Allergy. Clin. Immunol. 121, 1372–1378, 1378 e1371–1373 (2008).
Chanez, P. et al. Airway macrophages from patients with asthma do not proliferate. J. Allergy. Clin. Immunol. 92, 869–877 (1993).
Vissers, J.L., van Esch, B.C., Hofman, G.A. & van Oosterhout, A.J. Macrophages induce an allergen-specific and long-term suppression in a mouse asthma model. Eur. Respir. J. 26, 1040–1046 (2005).
Valstar, D.L. et al. Alveolar macrophages suppress non-specific inflammation caused by inhalation challenge with trimellitic anhydride conjugated to albumin. Arch. Toxicol. 80, 561–571 (2006).
Careau, E. et al. Antigen sensitization modulates alveolar macrophage functions in an asthma model. Am. J. Physiol. Lung. Cell. Mol. Physiol. 290, L871–879 (2006).
Holt, P.G. et al. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J. Exp. Med. 177, 397–407 (1993).
Upham, J.W., Strickland, D.H., Bilyk, N., Robinson, B.W. & Holt, P.G. Alveolar macrophages from humans and rodents selectively inhibit T-cell proliferation but permit T-cell activation and cytokine secretion. Immunology 84, 142–147 (1995).
Spiteri, M.A., Knight, R.A., Jeremy, J.Y., Barnes, P.J. & Chung, K.F. Alveolar macrophage-induced suppression of peripheral blood mononuclear cell responsiveness is reversed by in vitro allergen exposure in bronchial asthma. Eur. Respir. J. 7, 1431–1438 (1994).
Bilyk, N. & Holt, P.G. Inhibition of the immunosuppressive activity of resident pulmonary alveolar macrophages by granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 177, 1773–1777 (1993).
Balbo, P., Silvestri, M., Rossi, G.A., Crimi, E. & Burastero, S.E. Differential role of CD80 and CD86 on alveolar macrophages in the presentation of allergen to T lymphocytes in asthma. Clin. Exp. Allergy. 31, 625–636 (2001).
Burastero, S.E. et al. Increased expression of the CD80 accessory molecule by alveolar macrophages in asthmatic subjects and its functional involvement in allergen presentation to autologous TH2 lymphocytes. J. Allergy. Clin. Immunol. 103, 1136–1142 (1999).
Magnan, A., van Pee, D., Bongrand, P. & Vervloet, D. Alveolar macrophage interleukin (IL)-10 and IL-12 production in atopic asthma. Allergy 53, 1092–1095 (1998).
Plummeridge, M.J., Armstrong, L., Birchall, M.A. & Millar, A.B. Reduced production of interleukin 12 by interferon gamma primed alveolar macrophages from atopic asthmatic subjects. Thorax 55, 842–847 (2000).
Thomassen, M.J. et al. Nitric oxide inhibits inflammatory cytokine production by human alveolar macrophages. Am. J. Respir. Cell. Mol. Biol. 17, 279–283 (1997).
Song, C. et al. IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. J. Immunol. 181, 6117–6124 (2008).
Nomura, H. et al. Histamine stimulates alveolar macrophages to release neutrophil and monocyte chemotactic activity. J. Lab. Clin. Med. 138, 226–235 (2001).
Simon, H.U. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol. Rev. 193, 101–110 (2003).
Rubins, J.B. Alveolar macrophages: wielding the double-edged sword of inflammation. Am. J. Respir. Crit. Care. Med. 167, 103–104 (2003).
Alexis, N.E., Soukup, J., Nierkens, S. & Becker, S. Association between airway hyperreactivity and bronchial macrophage dysfunction in individuals with mild asthma. Am. J. Physiol. Lung. Cell. Mol. Physiol. 280, L369–L375 (2001).
Gosset, P. et al. Production of chemokines and proinflammatory and antiinflammatory cytokines by human alveolar macrophages activated by IgE receptors. J. Allergy. Clin. Immunol. 103, 289–297 (1999).
Fuller, R.W. et al. Immunoglobulin E-dependent stimulation of human alveolar macrophages: significance in type 1 hypersensitivity. Clin. Exp. Immunol. 65, 416–426 (1986).
Tamaoki, J., Sakai, N., Kanemura, T., Yamawaki, I. & Takizawa, T. IgE-dependent activation of alveolar macrophages augments neurally mediated contraction of small airways. Br. J. Pharmacol. 103, 1458–1462 (1991).
Rankin, J.A. et al. IgE-dependent release of leukotriene C4 from alveolar macrophages. Nature 297, 329–331 (1982).
Dery, R.E. & Bissonnette, E.Y. IFN-gamma potentiates the release of TNF-alpha and MIP-1alpha by alveolar macrophages during allergic reactions. Am. J. Respir. Cell. Mol. Biol. 20, 407–412 (1999).
Lohmann-Matthes, M.L., Steinmuller, C. & Franke-Ullmann, G. Pulmonary macrophages. Eur. Respir. J. 7, 1678–1689 (1994).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflicts of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Balhara, J., Gounni, A. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol 5, 605–609 (2012). https://doi.org/10.1038/mi.2012.74
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2012.74
This article is cited by
-
RNA m6A methylation modulates airway inflammation in allergic asthma via PTX3-dependent macrophage homeostasis
Nature Communications (2023)
-
OR2AT4 and OR1A2 counterregulate molecular pathophysiological processes of steroid-resistant inflammatory lung diseases in human alveolar macrophages
Molecular Medicine (2022)
-
The kinase p38α functions in dendritic cells to regulate Th2-cell differentiation and allergic inflammation
Cellular & Molecular Immunology (2022)
-
Smooth-muscle-derived WNT5A augments allergen-induced airway remodelling and Th2 type inflammation
Scientific Reports (2020)
-
Current E-Cigarette Research in the Context of Asthma
Current Allergy and Asthma Reports (2020)
