
Abstract
Epidemiological data indicate a link between rheumatoid arthritis (RA) and periodontal disease (PD). In vitro and in vivo studies have sought to dissect potential mechanisms by which PD may contribute to initiation and progression of RA. However, these are both multifactorial, chronic diseases, and their complex etiologies and pathogenesis themselves remain incompletely understood. Could there really be an etiological link or does this simply represent a statistical coincidence muddied by common risk factors? This review seeks to provide background on these two diseases in the context of recent discoveries suggesting that their pathogenesis may be related. In particular, the process of citrullination, a post-translational protein modification, has been highlighted as a process common to both diseases. The evidence for a relationship between the diseases is explored and its potential mechanisms discussed.
Similar content being viewed by others

Periodontal Disease
Periodontal disease (PD) is the most common cause of bone loss and infectious inflammatory disease of mankind, with mild forms of the disease affecting the majority of population and a more severe disease affecting 5–8%.1, 2, 3 PDs can be broadly classified as aggressive or chronic. The former affects younger patients and progresses rapidly, the latter, more common form, progresses relatively slowly and generally affects patients over 35 years of age. There are numerous subtle differences in etiology and pathogenesis of the two forms.4 In the main, this review describes studies of chronic periodontitis. The disease is clinically a readily accessed and visualized example of inflammation at a mucosal surface, which in a susceptible host leads to bone loss around the teeth and ultimately tooth loss (Figure 1). In addition, there is substantial evidence suggesting that patients with PD have between 1.2- and 6-fold increased risk of cardiovascular diseases, suggesting that inflammation at this mucosal surface has systemic consequences.5 This relationship shows a dose response, thus the greater the burden of PD, the greater the systemic impact. The microbial plaque biofilm is a necessary, but not a sufficient, cause of PD. Thus, variation in host response to the biofilm is believed to account for a significant proportion of the variation in disease susceptibility.6, 7, 8 The microbiology of periodontitis is complex, and most simplistically associated with a microbial community shift from a predominantly Gram-positive, aerobic health-associated biofilm, to a dominance of Gram-negative anaerobes. Seminal molecular studies have identified complexes of different bacterial species associated with severe chronic periodontitis, in particular Porphyromonas gingivalis, Tanerella forsythia, and Treponema denticola.9 More recently, sequencing studies unraveling the oral microbiome have served in part to confirm these findings and implicated other previously unidentified and highly fastidious species, such as Filifactor alocis, which significantly associate with disease.10 P. gingivalis has long been identified as a “periodontal pathogen”, as a species that is detected infrequently and in low numbers in health, and in greater frequency and larger numbers in destructive forms of the disease.11 In animal models, P. gingivalis serves as a “keystone pathogen” capable of instructing the development of a pathogenic flora, ultimately remaining in the matured microbial community only in small numbers.12 P. gingivalis has an impressive armamentarium of virulence factors including fimbrae, degradative enzymes, exopolysaccharide capsule, and atypical lipopolysaccharide (reviewed in detail by Holt et al.13). A group of extracellular cysteine proteases with trypsin-like proteolytic activity, known as gingipains, are associated with tissue damage and immune disruption in PD.14, 15, 16 Gingipains cleave host proteins at arginine or lysine residues by means of arg-gingipain A (RgpA) or B (RgpB), and lys-gingipain (Kgp), respectively.17 These enzymes have a critical role in P. gingivalis-mediated disease pathogenesis as evidenced by the attenuated virulence exhibited by gingipain knockout P. gingivalis.18 More recently, P. gingivalis has been identified as the only human parasitic prokaryotic organism expressing a peptidylarginine deiminase (PAD)—P. gingivalis peptidylarginine deiminase (PPAD), and is therefore capable of citrullinating proteins.19, 20

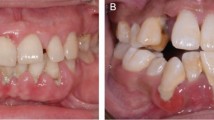
Aggressive periodontal disease causing bone loss around the teeth and loss of the upper front teeth in an otherwise healthy 33-year-old patient with a good standard of tooth cleaning. Periodontal disease does not always simply reflect oral neglect, and the variable response to the microbial plaque biofilm causes marked individual variation in disease susceptibility.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic autoimmune disease encompassing breach of self-tolerance, chronic inflammation, and joint destruction. RA affects ∼1% of the population and is associated with debilitating joint destruction and significant comorbidities including significantly accelerated atherosclerosis, psychological disease, and socioeconomic decline.21 Rheumatoid factor provides a serological indicator for RA, although offers only moderate disease sensitivity and specificity. The discovery of anti-citrullinated protein antibodies (ACPAs), as more sensitive and specific markers of RA, stemmed from observations that anti-keratin autoantibodies could be detected in 40–55% of RA patients,22 of which 40–50% tested negative for serum rheumatoid factor.23 These anti-keratin antibodies recognized an epitope containing citrulline, the deiminated form of arginine.24 Assessing IgG antibodies directed against a synthetic peptide containing citrulline, “Cyclic Citrullinated Peptide” is superior to either anti-keratin antibody or rheumatoid factor for diagnosis of RA.25 These ACPAs are present in 70% of RA patients, are associated with disease progression, and may be detectable up to 10 years before the clinical onset of disease.26, 27, 28, 29 IgA ACPA have been described and perhaps incriminate mucosal tissues in the development of autoimmunity.30 The role of ACPA and the numerous other contributors to the pathogenesis of RA has been extensively and elegantly reviewed elsewhere.30, 31, 32 RA disease associations with ACPAs have stimulated considerable interest in post-translationally modified proteins as sources of autoantigens. Citrullinated proteins may be created by peptidyl arginine deiminase enzymes (PAD), which substitute an amine residue on arginine with an oxygen group to form citrulline. More recently, a role in arthritis has also been proposed for homocitrullination (or carbamylation), an enzyme-independent reaction substituting an amine group of lysine with an oxygen group.33, 34, 35 In patients with erosive RA, carbamylated proteins and anti-carbamylated peptide antibodies are elevated compared with both healthy controls and non-erosive arthritis.36, 37
Periodontal destruction and the articular destruction of RA share common pathogenic mechanisms of host response-driven hard tissue destruction arising from chronic dysregulated inflammation in adjacent tissues. There is evidence that these diseases influence each other, a relationship potentially mediated by citrullination.
Associations between RA and PD
The concept of a relationship between periodontitis and RA was first mooted nearly 50 years ago38 and their common inflammatory mechanisms have been highlighted in numerous publications.39 Several studies demonstrate an epidemiological association between RA and PD. Patients with PD seem to have an increased likelihood of suffering RA.40, 41, 42 Conversely, RA patients show increased likelihood of PD, a relationship that could not be attributed simply to inadequate tooth cleaning in RA patients.43, 44, 45, 46 Analysis of the NHANES (National Health and Nutrition Examination Survey) showed that particular groups of RA patients were four times more likely to have PD than were those without RA.47 In another group, when compared with patients with osteoarthritis, RA patients also had significantly more PD. Moreover, in this group, ACPA titers correlated with PD severity.48 Among others, a hypothesis was proposed that periodontitis and exposure to P. gingivalis and its PPAD could predispose to autoimmunity to citrullinated proteins.49
There are associations between the humoral response to periodontal pathogens and RA. Compared with healthy controls, RA patients demonstrated significantly elevated anti-P. gingivalis IgG, and antibodies specific for other periodontal pathogens including Prevotella intermedia, Prevotella melaninogenica, and Tannerella forsythia. Elevated anti-P. gingivalis antibody titers were associated with higher serum concentrations of C-reactive protein and ACPA.49, 50, 51 A comparison of ACPA positive non-RA relatives with ACPA-positive RA patients, found that anti-P. gingivalis levels were significantly higher in the RA group.51 However, such cross-sectional studies cannot determine whether P. gingivalis infection precedes development of ACPA, and although there is convincing evidence of an ACPA–P. gingivalis association, in such studies, P. gingivalis antibody titers may be a surrogate marker for an independent, co-colonizing periodontal pathogen(s) or simply an indication of generalized periodontal inflammation. Furthermore, a direct comparison of the results of these studies is hindered by a lack of consensus methodology for quantifying PD,52 and it is tempting to speculate that the use of standardized periodontal microbiological assessments combined with methods to estimate the surface area of inflamed, ulcerated periodontal pocket epithelium may help clarify these findings.53
The associations revealed by the above studies, although intriguing, do not offer a cause and effect relationship. Nonetheless, there are experimental data suggesting that periodontitis and RA influence each other's pathogenesis.
Three small clinical trials have evaluated the possible therapeutic effects of periodontal treatment on symptoms and clinical markers of RA. RA patients received either comprehensive periodontal treatment, including removal of plaque biofilm from above and below the gum margin and oral hygiene instruction, or only basic periodontal treatment consisting of oral hygiene instruction and removal of plaque biofilm from above the gum only. Those receiving comprehensive treatment exhibited significantly improved erythrocyte sedimentation rate and health assessment questionnaire scores compared with the control group of RA patients who received only basic periodontal treatment.54 Periodontal treatment has been reported to lead to an improvement in disease activity scores (DAS28),55 and in a reduction in serum tumor necrosis factor-alpha (TNFα).56 Two small clinical studies have addressed the effects of treating arthritis on PD. Periodontal status was monitored in two groups of RA patients; one group received anti-TNFα treatment over the preceding 2 years, the other group was observed as anti-TNFα treatment commenced. Anti-TNFα treatment seemed to reduce bone destruction around the teeth. However, anti-TNFα treatment intriguingly seemed to cause more local gingival inflammation, highlighting that in the periodontium, inflammation and local destruction are related but are separate components of PD.57 A second study indicated that anti-TNFα treatment reduced both inflammation and local destruction.58 Although suggestive that reducing the inflammatory burden in the oral cavity is beneficial to patients with RA, and vice versa, these are small studies, and do not offer any mechanistic explanation of periodontal therapy ameliorating RA. Animal model studies have provided further detail and confirmation of these findings.
Rats challenged by subcutaneous implantation of sponges soaked with heat-killed P. gingivalis developed a more severe adjuvant-induced arthritis59 than did controls. A more subtle approach was subsequently adopted in which experimental PD was induced in mice by oral infection with P. gingivalis. Antibody-induced arthritis was then initiated, demonstrating that P. gingivalis-induced PD significantly exacerbated joint inflammation and bone destruction. Moreover, antibody-induced arthritis alone caused some periodontal destruction in the murine model,60 findings that are mirrored in other rodent models. Histological analysis of the mandibles of collagen-induced arthritis mice demonstrated severe periodontal destruction and alveolar bone loss.61 Similarly, marked periodontal destruction has been described in animals with antigen-induced arthritis,62 which was identified as dependent on TNFα and the oral microbiota. Similar findings were implied by earlier studies in which matrix metalloproteinase inhibition in a rat model of adjuvant arthritis eliminated the associated periodontal destruction.63 These in vivo studies suggest a possible bidirectional relationship between experimental arthritis and PD, suggesting that bone destruction in these models is not exclusive to synovial joints, and that the oral flora is required for the associated bone destruction.
Below, some potential mechanisms by which PD may influence arthritis are explored (Figure 2). This can be viewed in many ways, and for ease of discussion, are broadly grouped here as (i) bacterial generation of autoantigens, (ii) local inflammation generating autoantigens, (iii) direct bacterial insult, and (iv) systemic inflammatory effects of PD. These pathways exist against a backdrop-shared genetic, environmental, and behavioral risk factors, which are discussed separately. Clearly, the list is not exhaustive, and the pathways are not mutually exclusive.

Schematic representation of mechanisms by which periodontal inflammation may affect rheumatoid arthritis. Each could hypothetically occur in isolation but more likely in combination, and always against a background of common risk factors.
Bacterial Generation of Autoantigens
Citrullination by bacterial enzymes
As noted, the periodontal pathogen P. gingivalis is capable of citrullinating both its own and its host proteins. P. gingivalis gingipains create substrate for PPAD by cleaving at arginine residues, suggesting a system integral to this species’ metabolism. The physiological role of PPAD remains uncertain, although it has been shown to inactivate hemagglutinins, possibly furnishing P. gingivalis with a continued supply of nutrients from the vascular system.64 PPAD may assist with bacterial defenses against neutrophils and neutralize acidic environments through the production of ammonia. PPAD differs from human PAD in its structure, its ability to citrullinate-free arginine and function independently of calcium concentration.19, 64 In vitro, P. gingivalis gingipains degrade human fibrinogen and α-enolase, which are then citrullinated by PPAD.20 It has been proposed that P. gingivalis may have an indirect role in promoting human PAD activity by increasing intracellular calcium concentrations through induction of defensive enzymatic activity.64, 65
Citrullinated bacterial proteins as autoantigens
In addition to citrullinating host proteins, bacterial citrullination of bacterial proteins provides a potential source of autoantigens. Bacterial α-enolase is a candidate autoantigen, with significant homology between human and bacterial α-enolase. α-Enolase was found to be reactive with sera from 25% of RA patients in a study of 255 very-early-disease-onset RA patients, and was predictive of severe radiological disease progression in this cohort.66 Subsequent studies narrowed this reactivity to citrullinated α-enolase67 after which anti-α-enolase antibodies were detected in ACPA-positive subgroups of three cohorts of Northern European RA patients (43–63%).68 Further investigation demonstrated significantly elevated levels of α-enolase in the synovial joints of patients with RA and spondylarthridities compared with the joints of osteoarthritis patients. Anti-α-enolase antibodies were not detected in the synovium of spondylarthridities patients, but were identified in 12 of the 20 RA patients.69 Autoantibodies directed against citrullinated α-enolase were predictive of RA, with 46% of RA sera recognizing the protein in its citrullinated form compared with 13% when non-citrullinated.67 Immunological mapping using a library of cyclic citrullinated α-enolase peptides led to the identification of a B cell-dominant epitope comprising amino acids 5–21 of α-enolase (5KIHAREIFDSRGNPTVE21) where arginine-9 and arginine-15 are citrullinated. This peptide, “citrullinated enolase peptide-1” (CEP-1) is highly conserved in prokaryotes and eukaryotes. Indeed, at the CEP-1 region, human α-enolase has an 82% sequence similarity with that of P. gingivalis, whereas a 9 amino-acid span including arginine-15 of CEP-1 is 100% identical.70, 71 Immunization with citrullinated human and P. gingivalis α-enolase,72 and citrullinated fibrinogen73 cause similar pathology in humanized DR4 transgenic mice. These findings suggest that by mimicking the molecular structure of host-citrullinated proteins, PPAD-citrullinated bacterial α-enolase could trigger a loss of tolerance to structurally similar host proteins, resulting in expression of ACPA, and the development of RA.
Local Inflammation Generating Autoantigens
Neutrophil extracellular traps (NETs) are extracellular fibrous webs that bind Gram-negative bacteria, Gram-positive bacteria, and fungi.74, 75 NET formation requires histone citrullination for the decondensation and unfolding of chromatin, and the formation of the web-like structure of NETs.76 Citrullination is necessary for NET formation and dependent on neutrophil-derived PAD4,76 confirmed by the lack of NET formation and deficiency in bacterial killing in PAD4 knockout mice.77, 78 NET formation has been observed in PD, both by human studies and in vitro models, and these studies suggest that the entrapment and flushing of bacteria may be more important in NET activity than bacterial killing.79 The bacterially infected periodontal tissues are infiltrated with activated neutrophils and are therefore likely to be exposed to higher concentrations of neutrophil-expressed PAD4. Chronic PAD4 exposure may lead to increased host peptide citrullination and breakdown of immune tolerance in an RA-susceptible individual. As such, perhaps either singly or in combination, NET-related citrullination and PPAD activity could prime an autoimmune response in a susceptible individual.
The oral cavity is a potential source of homocitrullinated (carbamylated) proteins and there is compelling recent evidence of a key role for carbamylated host proteins in the pathology of RA.36, 37 Homocitrulline is generated from lysine in an enzyme-independent reaction involving urea-derived cyanate, in which the amine group of lysine is substituted with an oxygen group. The normal physiological concentrations of urea and cyanate are considered too low to permit extensive homocitrullination of proteins. However, in inflammation, myeloperoxidase released from neutrophils may convert thiocyanate into cyanate and thus increase the potential for carbamylation of proteins.80 As a result of neutrophil infiltration, myeloperoxidase levels are increased in individuals with PD, correlate positively with disease severity, and decrease after successful clinical intervention.81, 82, 83 Hypothetically, myeloperoxidase-mediated increased conversion of salivary thiocyanate to cyanate may cause an increase in carbamylation of proteins. Although salivary and plasma thiocyanate levels are increased in smokers and inflammation,80, 84 studies investigating salivary thiocyanate or cyanate levels in PD sufferers were not identified at this time. Moreover, carbamylation of collagen readily occurred in vivo after neutrophil activation85 and is linked to matrix metalloproteinase release.86 As such, in addition to bacteria-related citrullination, the inflammation in the oral cavity resulting from periodontitis is a potential cause of post-translational protein modification and autoantigen generation.
Direct Bacterial Insult
P. gingivalis is invasive, with a capability to journey from one cell type to another.87, 88 In addition, P. gingivalis, or its components, may translocate from the oral cavity to distant sites during bacteremias known to occur during eating and tooth cleaning.89 Therefore, theoretically bacteria and/or components may, by chance or sequestration, migrate to the joint and be processed by local antigen-presenting cells in the synovium. P. gingivalis and other periodontopathic bacteria have been epidemiologically linked to various other systemic conditions including atherosclerosis.90, 91 Indeed, P. gingivalis DNA has been identified in atherosclerotic plaques and vessels.91, 92, 93, 94 P. gingivalis DNA was detected in the synovial fluid of up to 80% of RA patients while being absent from that of healthy individuals.95, 96 Viable P. gingivalis has not been detected in the synovium. Original findings of P. gingivalis DNA in atherosclerotic plaques were only later extended by bacterial culture to reveal viable bacteria,97 although the strict growth requirements of these bacteria seem likely to preclude their colonization of an environment as foreign as the synovium. It remains unclear whether preexisting joint inflammation results in bacterial sequestration to the synovium or if bacterial products initiate inflammation in the joint and subsequent pathogenesis of RA. Irrespective, the inflamed synovium is likely to act as a non-specific trap for bacterial fragments, and these trapped fragments may in turn elicit an inflammatory response thus perpetuating the existing lesion. Activation of Toll-like receptors (TLRs) by pathogen-associated molecular patterns has been implicated in the pathogenesis of RA,98 and expression of TLR2, TLR3, TLR4, TLR6, TLR7, and TLR9 has been demonstrated in the rheumatoid joint, on macrophages and fibroblast-like synoviocyte lineages. No association with TLR polymorphisms has been detected, but in vivo studies of gene knock-outs, in particular TLR2−/− and TLR4−/−, clearly document their role in initiating and exacerbating arthritis (reviewed in the study by Drexler and Foxwell99). P. gingivalis lipopolysaccharide is atypical in that it activates host innate immune responses through stimulation of TLR2 and TLR4,100 and thus P. gingivalis sequestration in the inflamed synovium could further exacerbate inflammation in the joint through TLR2 and TLR4. P. gingivalis DNA mediates TLR9-driven inflammatory cytokine production.101 At this time, it is difficult to delineate the contribution of P. gingivalis from other bacterial species and danger-associated molecular patterns that could elicit a similar response. It would be interesting to explore pathogen-associated molecular pattern and P. gingivalis antigen expression in the joints of a cohort of PD/RA patients. In addition to bacterial agonists of TLRs, it could be hypothesized that PPAD-citrullinated P. gingivalis or self proteins may exacerbate pathology through TLR engagement leading to increased TNF-α production. Citrullinated fibrinogen has been shown to increase TNF-α production by synovial macrophages through co-stimulation of TLR-4 and FcγR.102 The role of enolase and other citrullinated P. gingivalis proteins as inducers of TLR-driven TNF-α production is an intriguing avenue for further studies investigating PD and RA.
PD lesions offer a reservoir of potentially autoantigenic-citrullinated proteins, and components of P. gingivalis can translocate to the joint. Whether P. gingivalis-citrullinated proteins can ultimately breach tolerance, either locally in the oral cavity, systemically, or localizing to the joint, has not been established.
Systemic Inflammatory Effects of Periodontits
Advanced and aggressive forms of PD demonstrate evidence of inflicting a systemic inflammatory burden, which can be reduced by periodontal treatment, as assessed by changes in serum C-reactive protein and interleukin-6.103 Elevated interleukin-17A has been reported in serum of patients with aggressive periodontitis,104 and although Th17 cells have been identified locally in the inflamed periodontium,105 there is less certainty that circulating T-cell populations are influenced by PD. However, influences on other populations of circulating leukocytes are apparent. P. gingivalis may enter and remain viable in circulating dendritic cells and promote a semi-matured, highly migratory phenotype.88, 106 A substantial (up to a five-fold) increase in circulating CD19+ CD5+ B cells was observed in patients with periodontitis compared with healthy controls.107 Moreover, circulating B cells in patients with aggressive PD demonstrated intriguing functional discrepancies, with an elevated proportion of B cells expressing TLR2, with a hyperresponsiveness to TLR2 and TLR4 ligands in vitro.108 Therefore, PD could influence autoimmunity by creating a systemic inflammatory milieu conducive to both development of autoimmunity and progression to clinical autoimmune disease in a susceptible individual. Given the success of T-cell, B-cell, and cytokine-targeted therapies in RA,109 it seems intuitive that periodontal inflammation could at the very least exacerbate RA by fuelling these inflammatory pathways. Dissecting this generalized dysregulated inflammation is complicated by the multifactorial etiology and common risk factors of both PD and RA.
Shared Risk Factors
Genetic
The “shared epitope” (SE) describes human leukocyte antigen (HLA) molecules including one of three amino-acid sequences at position 70–74 of the DR chain. This “shared epitope” significantly predisposes to ACPA-positive RA (reviewed in the study by Bax et al.110). To date, evidence linking HLA-DR-SEs to periodontitis is less conclusive. However, there is a suggestion of increased susceptibility to periodontitis in individuals expressing HLA-DR4, with this allele expressed in 80% of rapidly progressing periodontitis patients and 38% of healthy controls.111 Some studies show associations with the HLA-DRB1*04 haplotype,112 in PD patients,113, 114, 115, 116 although others show no correlation.117, 118, 119 Meta-analysis of numerous studies investigating HLA alleles in periodontitis suggested no significant associations with HLA-DR4, or any other class II antigens, but associations with HLA-A19 and HLA-B15.120 At present, larger studies would be required to draw conclusions about the relationship between HLA-DR4 and periodontitis.
Smoking–gene interactions
Smoking is a well-established risk factor for PD, with up to 50% of cases attributable to current or former smoking and a clear dose–response relationship between disease severity and number of cigarettes smoked.121 Moreover, smokers respond significantly less favorably to periodontal treatment.122 P. gingivalis has been found in significantly higher levels in the periodontium of smokers compared with non-smokers123, 124 possibly as a result of the deleterious effect smoking has on the inflammatory capabilities of the periodontal tissues.125
Smoking is a well-established risk factor for RA (reviewed in the study by Klareskog et al.126), and studies of the smoking–RA relationship demonstrate a gene environment–behavior interaction. Citrulline positivity was detected in 13.75% of bronchoalveolar lavage cells of non-smokers compared with 28.5% in smokers,127 with higher bronchial and alveolar PAD2 expression in smokers compared with non-smokers,128 therefore creating a source of citrullinated autoantigen. The Epidemiological Investigation of Rheumatoid Arthritis case–control study demonstrated an association between SE alleles and smoking.129 Further studies confirmed a strong association between smoking and ACPA-positive RA,127, 130, 131 whereas a similar relationship was not observed in ACPA-negative RA.132 The smoking–gene interaction demonstrated a dose-response gradient in some studies133, 134, 135 with a 40-fold increased risk of disease in ACPA-positive heavy smokers who were homozygous for the SE.134 However, there were contradictory results in an African-American cohort, which may reflect differences in both ethnicity and environmental exposures, which may mediate the association between ACPA-negative RA and smoking.136 Other studies found conflicting evidence of the influence of smoking on RA disease severity137, 138, 139 and some have found little evidence of a strong gene–environment interaction in the development of RA.140 Explanations for the discrepant findings include variations in the procedures for recruiting controls and patients, differing methods of quantifying smoking habits, and existence of environmental exposures.132 This last factor may prove the most intriguing. Air pollution was suggested as a potential confounder, which could cause increased lung PAD activity. To date, none of these studies have included periodontal status or investigated P. gingivalis infection, and this may unravel some of these apparently discrepant findings. Regional variation in periodontal infection has been investigated by comparing the microbiota in periodontal pockets of groups of periodontitis subjects from Brazil, Chile, Sweden, USA, and Greece. After adjustment for age, PD severity, gender, and smoking, a three-fold increase in the mean percentage of P. gingivalis DNA in the US sample was observed compared with the Swedish sample.141 Furthermore, different clonal types of P. gingivalis were found in healthy and periodontitis subjects, highlighting that pathogens show strain diversity, which may result in a variation in expression of virulence factors, which may affect the local burden of citrullinated proteins. Therefore, it is possible variable rates of P. gingivalis infection as well as differences in the strains of P. gingivalis involved could further confound the smoking/RA studies.
Thus, long-term cigarette smoke exposure in the lungs results in a chronic inflammatory response, which in turn upregulates peptidylarginine deiminase expression by macrophages within the pulmonary architecture, leading to increased PAD activity and citrullinated peptide production. Smoking causes an increase not just in pulmonary PAD2 activity but also in periodontal P. gingivalis colonization and, presumably, PPAD production, therefore potentially initiating two separate first “hits” in two different areas of the body.
The “Two-Hit” RA Pathogenesis Model—RA-Smoking Hypothesis
On the basis of the above findings in smokers, a “two-hit” hypothesis was proposed to explain the development of RA in smokers. The first “hit” in this model is initiated by cigarette smoking which, as described above, leads to increased citrullinated proteins. Loss of immune tolerance to citrullinated peptides such as citrullinated fibrinogen,142 vimentin,143 and type II collagen144 in susceptible individuals results in the production of ACPA. Possibly years after this initial priming of the immune system, a second “hit” occurs. From this hypothesis, it is unclear why autoimmunity to citrullinated peptides is associated with RA but not other organ-specific autoimmune conditions, although it has been observed that synovial joints are particularly sensitive to inflammatory stimuli.145 An undefined synovial inflammation could lead to synovial citrullinated protein production, a subsequent autoimmune response, and finally RA. Arguably, PD may, similar to cigarette smoking, offer a “first hit.”
Conclusion: How are PD and RA Related?
The human cross-sectional studies allow a “snap shot” of the RA–PD association at one specific point in time and are therefore unable to provide reliable evidence on causality. The animal studies suggest that the diseases influence each other's pathogenesis. At present, it could be suggested that RA causes PD, PD causes RA, or neither disease causes the other, but both diseases are caused independently by a third factor. None of the evidence as yet substantially indicates that PD causes RA, although the proposed mechanisms that link the two diseases provide attractive explanations for many of the observations. In the authors’ opinion, it will be key to identify how and when PD fits in the pathway to RA and whether this can be modulated. Manipulations of the pathogenic bacteria and the host immune system during experimental disease models will offer significant insight and guide clinical studies. Combinations of human and animal studies will ultimately unravel whether systemic inflammation, direct bacterial insult, citullinated autoantigens, or other mechanisms mediate the PD–RA relationship. Assuming a causal relationship exists, it is improbable that only one of these mechanisms is solely responsible, and in an RA–PD patient, it is conceivable that two or more mechanisms combine to mediate the relationship. Nonetheless, understanding such a relationship seems a promising avenue to yield improved therapeutic strategies—potentially for both diseases independently and in combination.
References
Loe, H., Anerud, A., Boysen, H. & Morrison, E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. J. Clin. Periodontol. 13, 431–445 (1986).
Hugoson, A., Sjödin, B. & Norderyd, O. Trends over 30 years, 1973–2003, in the prevalence and severity of periodontal disease. J. Clin. Periodontol. 35, 405–414 (2008).
Morris, A.J., Steele, J. & White, D.A. Adult dental health survey: the oral cleanliness and periodontal health of UK adults in 1998. Br. Dent. J. 191, 186–192 (2001).
Armitage, G.C. Periodontal diagnoses and classification of periodontal diseases. Periodontol. 2000 34, 9–21 (2004).
Kebschull, M., Demmer, R.T. & Papapanou, P.N. Gum bug, leave my heart alone! Epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J. Dent. Res. 89, 879–902 (2010).
Trombone, A.P. et al. Periodontitis and arthritis interaction in mice involves a shared hyper-inflammatory genotype and functional immunological interferences. Genes Immun. 11, 479–489 (2010).
Kinane, D.F. & Mark Bartold, P. Clinical relevance of the host responses of periodontitis. Periodontol. 2000 43, 278–293 (2007).
Berglundh, T. & Donati, M. Aspects of adaptive host response in periodontitis. J. Clin. Periodontol. 32 (Suppl 6), 87–107 (2005).
Socransky, S.S., Cugini, M.A., Smith, C. & Kent, R.L. Jr Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25, 134–144 (1998).
Kumar, P., Leys, E.J., Bryk, J.M., Martinez, F.J., Moeschberger, M.L. & Griffen, A.L. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J. Clin. Microbiol. 44, 3665–3673 (2006).
Socransky, S.S. & Haffajee, A.D. Effect of therapy on periodontal infections. J. Periodontol. 64 (8 Suppl), 754–759 (1993).
Hajishengallis, G. et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10, 497–506 (2011).
Holt, S.C., Kesavalu, L., Walker, S. & Genco, C.A. Virulence factors of Porphyromonas gingivalis. Periodontol. 2000 20, 168–238 (1999).
Kesavalu, L., Holt, S.C. & Ebersole, J.L. Trypsin-like protease activity of Porphyromonas gingivalis as a potential virulence factor in a murine lesion model. Microb. Pathog. 20, 1–10 (1996).
Birkedal-Hansen, H., Taylor, R.E., Zambon, J.J., Barwa, P.K. & Neiders, M.E. Characterization of collagenolytic activity from strains of Bacteroides gingivalis. J. Periodontal. Res. 23, 258–264 (1988).
Lamont, R.J. & Jenkinson, H.F. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62, 1244–1263 (1998).
Curtis, M.A., Aduse-Opoku, J. & Rangarajan, M. Cysteine proteases of Porphyromonas gingivalis. Crit. Rev. Oral Biol. Med. 12, 192–216 (2001).
Guo, Y., Nguyen, K-A. & Potempa, J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon's knife to a meat chopper-like brutal degradation of proteins. Periodontol. 2000 54, 15–44 (2010).
McGraw, W.T., Potempa, J., Farley, D. & Travis, J. Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infection Immunity 67, 3248–3256 (1999).
Wegner, N. et al. Peptidylarginine deiminase from porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 62, 2662–2672 (2010).
Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 423, 356–361 (2003).
Vincent, C. et al. High diagnostic value in rheumatoid arthritis of antibodies to the stratum corneum of rat oesophagus epithelium, so-called ‘antikeratin antibodies. Ann. Rheum. Dis. 48, 712–722 (1989).
Cordonnier, C. et al. Diagnostic value of anti-RA33 antibody, antikeratin antibody, antiperinuclear factor and antinuclear antibody in early rheumatoid arthritis: comparison with rheumatoid factor. Br. J. Rheumatol. 35, 620–624 (1996).
van Venrooij, W.J. & Pruijn, G.J. Citrullination: a small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res. 2, 249–251 (2000).
Schellekens, G.A. et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 43, 155–163 (2000).
Zendman, A.J.W., Vossenaar, E.R. & Van Venrooij, W.J. Autoantibodies to citrullinated (poly)peptides: a key diagnostic and prognostic marker for rheumatoid arthritis. Autoimmunity 37, 295–299 (2004).
van Gaalen, F.A. et al. Association between HLA class II genes and autoantibodies to cyclic citrullinated peptides (CCPs) influences the severity of rheumatoid arthritis. Arthritis Rheum. 50, 2113–2121 (2004).
Meyer, O. et al. Anticitrullinated protein/pepticle antibody assays in early rheumatoid arthritis for predicting five year radiographic damage. Ann. Rheum. Dis. 62, 120–126 (2003).
Nielen, M.M.J. et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis—a study of serial measurements in blood donors. Arthritis Rheum. 50, 380–386 (2004).
Klareskog, L., Ronnelid, J., Lundberg, K., Padyukov, L. & Alfredsson, L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu. Rev. Immunol. 26, 651–675 (2008).
van Gaalen, F., Ioan-Facsinay, A., Huizinga, T.W.J. & Toes, R.E.M. The devil in the details: the emerging role of anticitrulline autoimmunity in rheumatoid arthritis. J. Immunol. 175, 5575–5580 (2005).
Suwannalai, P., Trouw, L.A., Toes, R.E. & Huizinga, T.W. Anti-citrullinated protein antibodies (ACPA) in early rheumatoid arthritis. Mod. Rheumatol. Published online 6 July 2011.
Erill, S., Calvo, R. & Carlos, R. Plasma protein carbamylation and decreased acidic drug protein binding in uremia. Clin. Pharmacol. Ther. 27, 612–618 (1980).
Fluckiger, R., Harmon, W., Meier, W., Loo, S. & Gabbay, K.H. Hemoglobin carbamylation in uremia. N Engl. J. Med. 304, 823–827 (1981).
Van Lente, F., McHugh, A. & Pippenger, C.E. Carbamylation of apo-aspartate aminotransferase: a possible mechanism for enzyme inactivation in uremic patients. Clin. Chem. 32, 2107–2108 (1986).
Mydel, P. et al. Carbamylation-dependent activation of T cells: a novel mechanism in the pathogenesis of autoimmune arthritis. J. Immunol. 15, 6882–6890 (2010).
Shi, J. et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc. Natl Acad. Sci. USA 108, 17372–17377 (2011).
Liubomorova, I. State of periodontium in patients affected with rheumatism. Stomatologiia 43, 33–37 (1964).
Snyderman, R. & McCarty, G.A. Analogous mechanisms of tissue destruction in rheumatoid arthritis and periodontal disease. In (R.J. Genco SEM, ed) ( American Society of Microbiology, Washington, DC, 1982 ).
Mercado, F., Marshall, R.I., Klestov, A.C. & Bartold, P.M. Is there a relationship between rheumatoid arthritis and periodontal disease? J. Clin. Periodontol. 27, 267–272 (2000).
Mercado, F.B., Marshall, R.I., Klestov, A.C. & Bartold, P.M. Relationship between rheumatoid arthritis and periodontitis. J. Periodontol. 72, 779–787 (2001).
Arkema, E.V., Karlson, E.W. & Costenbader, K.H. A prospective study of periodontal disease and risk of rheumatoid arthritis. J. Rheumatol. 37, 1800–1804 (2010).
Abdelsalam, S.K., Hashim, N.T., Elsalamabi, E.M. & Gismalla, B.G. Periodontal status of rheumatoid arthritis patients in Khartoum state. BMC Res. Notes 28, 460 (2011).
Gleissner, C., Willershausen, B., Kaesser, U. & Bolten, W.W. The role of risk factors for periodontal disease in patients with rheumatoid arthritis. Eur. J. Med. Res. 18, 387–392 (1998).
Kasser, U., Gleissner, C., Dehne, F., Michel, A., Willershausen-Zonnchen, B. & Bolten, W.W. Risk for periodontal disease in patients with longstanding rheumatoid arthritis. Arthritis. Rheum. 40, 2248–2251 (1997).
Cooles, F.A. & Isaacs, J.D. Pathophysiology of rheumatoid arthritis. Curr. Opin. Rheumatol. 23, 233–240 (2011).
de Pablo, P., Dietrich, T. & McAlindon, T.E. Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. J. Rheumatol. 35, 70–76 (2008).
Dissick, A. et al. Association of periodontitis with rheumatoid arthritis: a pilot study. J. Periodontol. 81, 223–230 (2010).
Rosenstein, E., Greenwald, R., Kushner, L. & Weissmann, G. Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation 28, 311–318 (2004).
Haffajee, A.D. & Socransky, S.S. Microbial etiological agents of destructive periodontal diseases. Periodontol. 2000 5, 78–111 (1994).
Hitchon, C.A. et al. Antibodies to porphyromonas gingivalis are associated with anticitrullinated protein antibodies in patients with rheumatoid arthritis and their relatives. J. Rheumatol. 37, 1105–1112 (2010).
Preshaw, P.M. Definitions of periodontal disease in research. J. Clin. Periodontol. 36, 1–2 (2009).
Nesse, W., Abbas, F., van der Ploeg, I., Spijkervet, F.K.L., Dijkstra, P.U. & Vissink, A. Periodontal inflamed surface area: quantifying inflammatory burden. J. Clin. Periodontol. 35, 668–673 (2008).
Ribeiro, J., Leao, A. & Novaes, A.B. Periodontal infection as a possible severity factor for rheumatoid arthritis. J. Clin. Periodontol. 32, 412–416 (2005).
Al-Katma, M.K., Bissada, N.F., Bordeaux, J.M., Sue, J. & Askari, A.D. Control of periodontal infection reduces the severity of active rheumatoid arthritis. J. Clin. Rheumatol. 13, 134–137 (2007).
Ortiz, P. et al. Periodontal therapy reduces the severity of active rheumatoid arthritis in patients treated with or without tumor necrosis factor inhibitors. J. Periodontol. 80, 535–540 (2009).
Pers, J.O., Saraux, A., Pierre, R. & Youinou, P. Anti-TNF-alpha immunotherapy is associated with increased gingival inflammation without clinical attachment loss in subjects with rheumatoid arthritis. J. Periodontol. 79, 1645–1651 (2008).
Mayer, Y., Balbir-Gurman, A. & Machtei, E.E. Anti-tumor necrosis factor-alpha therapy and periodontal parameters in patients with rheumatoid arthritis. J. Periodontol. 80, 1414–1420 (2009).
Bartold, P.M., Marino, V., Cantley, M. & Haynes, D.R. Effect of Porphyromonas gingivalis-induced inflammation on the development of rheumatoid arthritis. J. Clin. Periodontol. 37, 405–411 (2010).
Cantley, M.D., Haynes, D.R., Marino, V. & Bartold, P.M. Pre-existing periodontitis exacerbates experimental arthritis in a mouse model. J. Clin. Periodontol. 38, 532–541 (2011).
Park, J.C. et al. Mechanism of alveolar bone loss in a collagen-induced arthritis model in mice. J. Clin. Periodontol. 38, 122–130 (2011).
Queiroz-Junior, C.M. et al. Experimental arthritis triggers periodontal disease in mice: involvement of TNFα and the oral microbiota. J. Immunol. 187, 3821–3830 (2011).
Ramamurthy, H.S., Greenwald, R.A., Celiker, M.Y. & Shi, E.Y. Experimental arthritis in rats induces biomarkers of periodontitis which are ameliorated by gene therapy with tissue inhibitor of matrix metalloproteinases. J. Periodontol. 76, 229–233 (2005).
Mangat, P., Wegner, N., Venables, P.J. & Potempa, J. Bacterial and human peptidylarginine deiminases: targets for inhibiting the autoimmune response in rheumatoid arthritis?. Arthritis Res. Ther. 12, 209 (2010).
Rodriguez, S.B., Stitt, B.L. & Ash, D.E. Cysteine 351 is an essential nucleophile in catalysis by Porphyromonas gingivalis peptidylarginine deiminase. Arch. Biochem. Biophys. 504, 190–196 (2010).
Saulot, V. et al. Presence of autoantibodies to the glycolytic enzyme alpha-enolase in sera from patients with early rheumatoid arthritis. Arthritis Rheum. 46, 1196–1201 (2002).
Kinloch, A. et al. Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res. Ther. 7, R1421–R1429 (2005).
Mahdi, H. et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat. Genet. 41, 1319–1324 (2009).
Kinloch, A. et al. Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis. Arthritis Rheum. 58, 2287–2295 (2008).
Lundberg, K., Wegner, N., Yucel-Lindberg, T. & Venables, P.J. Periodontitis in RA the citrullinated enolase connection. Nat. Rev. Rheumatol. 6, 727–730 (2010).
Lundberg, K. et al. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 58, 3009–3019 (2008).
Kinloch, A.J. et al. Immunisation with Porphyromonas gingivalis enolase induces autoimmunity to mammalian alpha-enolase and arthritis in DR4-IE transgenic mice. Arthritis Rheum. 63, 3818–3823 (2011).
Hill, J.A. et al. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J. Exp. Med. 205, 967–979 (2008).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 5663, 1532–1535 (2004).
Brinkmann, V. & Zychlinsky, A. Beneficial suicide: why neutrophils die to make NETs. Nat. Rev. Microbiol. 5, 577–582 (2007).
Neeli, I., Khan, S.N. & Radic, M. Histone deimination as a response to inflammatory stimuli in neutrophils. J. Immunol. 180, 1895–1902 (2008).
Wang, Y.M. et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184, 205–213 (2009).
Li, P., Li, M., Lindberg, M.R., Kennett, M.J., Xiong, N. & Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862 (2010).
Vitkov, L., Klappacher, M., Hannig, M. & Krautgartner, W.D. Extracellular neutrophil traps in periodontitis. J. Periodontal. Res. 44, 664–672 (2009).
Sirpal, S. Myeloperoxidase-mediated lipoprotein carbamylation as a mechanistic pathway for atherosclerotic vascular disease. Clin. Sci. 116, 681–695 (2009).
Smith, Q.T., Hinrichs, J.E. & Melnyk, R.S. Gingival crevicular fluid myeloperoxidase at periodontitis sites. J. Periodontal. Res. 21, 45–55 (1986).
Cao, C.F. & Smith, Q.T. Crevicular fluid myeloperoxidase at healthy, gingivitis and periodontitis sites. J. Clin. Periodontol. 16, 17–20 (1989).
Buchmann, R., Hasilik, A., Van Dyke, T.E. & Lange, D.E. Resolution of crevicular fluid leukocyte activity in patients treated for aggressive periodontal disease. J. Periodontol. 73, 995–1002 (2002).
Wang, Z. et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 13, 1176–1184 (2007).
Jaisson, S., Delevallee-Forte, C., Toure, F., Rieu, P., Garnotel, R. & Gillery, P. Carbamylated albumin is a potent inhibitor of polymorphonuclear neutrophil respiratory burst. FEBS Lett. 581, 1509–1513 (2007).
Garnotel, R., Sabbah, N., Jaisson, S. & Gillery, P. Enhanced activation of and increased production of matrix metalloproteinase-9 by human blood monocytes upon adhering to carbamylated collagen. FEBS Lett. 563, 13–16 (2004).
Yilmaz, O., Verbeke, P., Lamont, R.J. & Ojcius, D.M. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect. Immun. 74, 703–710 (2006).
Carrion, J.A.G.Y. & Cutler, C. Pathogen trafficking dendritic cells in periodontitis: links to coronary artery disease IADR; San Diego, CA. J. Dent. Res. ( www.dentalresearch.org ). abstract number 2809 (2011).
Crasta, K., Mitchell, D., Curtis, B., Stewart, D. & Heitz-Mayfield, L.J.A. Bacteraemia due to dental flossing. J. Clin. Periodontol. 36, 323–332 (2009).
Pitiphat, W., Joshipura, K.J., Gillman, M.W., Williams, P.L., Douglass, C.W. & Rich-Edwards, J.W. Maternal periodontitis and adverse pregnancy outcomes. Community Dent. Oral Epidemiol. 36, 3–11 (2008).
Haraszthy, V.I., Zambon, J.J., Trevisan, M., Zeid, M. & Genco, R.J. Identification of periodontal pathogens in atheromatous plaques. J. Periodontol. 71, 1554–1560 (2000).
Fiehn, N.E., Larsen, T., Christiansen, N., Holmstrup, P. & Schroeder, T.V. Identification of periodontal pathogens in atherosclerotic vessels. J. Periodontol. 76, 731–736 (2005).
Padilla, C. et al. Periodontal pathogens in atheromatous plaques isolated from patients with chronic periodontitis. J. Periodontal. Res. 41, 350–353 (2006).
Romano, F., Barbui, A. & Aimetti, M. Periodontal pathogens in periodontal pockets and in carotid atheromatous plaques. Minerva Stomatol. 56, 169–179 (2007).
Martinez-Martinez, R.E., Abud-Mendoza, C., Patino-Marin, N., Rizo-Rodriguez, J.C., Little, J.W. & Loyola-Rodriguez, J.P. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J. Clin. Periodontol. 36, 1004–1010 (2009).
Moen, K. et al. Synovial inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral bacterial DNAs. Clin. Exp. Rheumatol. 24, 656–663 (2006).
Kozarov, E.V., Dorn, B.R., Shelburne, C.E., Dunn, W.A. Jr & Progulske-Fox, A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler. Thromb. Vasc. Biol. 25, 17–18 (2005).
van der Heijden, I.M. et al. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 43, 593–598 (2000).
Drexler, S.K. & Foxwell, B.M. The role of Toll-like receptors in chronic inflammation. Int. J. Biochem. Cell Biol. 42, 506–518 (2010).
Bainbridge, B.W., Coats, S.R. & Darveau, R.P. Porphyromonas gingivalis lipopolysaccharide displays functionally diverse interactions with the innate host defense system. Ann. Periodontol. 7, 29–37 (2002).
Sahingur, S.E., Xia, X.J., Alamgir, S., Honma, K., Sharma, A. & Schenkein, H.A. DNA from Porphyromonas gingivalis and Tannerella forsythia induce cytokine production in human monocytic cell lines. Mol. Oral Microbiol. 25, 23–35 (2010).
Sokolove, J., Zhao, X., Chandra, P.E. & Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum. 63, 53–62 (2011).
D'Aiuto, F. et al. Periodontitis and systemic inflammation: control of the local infection is associated with a reduction in serum inflammatory markers. J. Dent. Res. 83, 156–160 (2004).
Schenkein, H.A., Koertge, T.E., Brooks, C.N., Sabatini, R., Purkall, D.E. & Tew, J.G. IL-17 in Sera from patients with aggressive periodontitis. J. Dent. Res. 89, 943–947 (2010).
Cardoso, C.R. et al. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol. Immunol. 24, 1–6 (2009).
Zeituni, A.E., Carrion, J. & Cutler, C.W. Porphyromonas gingivalis-dendritic cell interactions: consequences for coronary artery disease. J. Oral. Microbiol. Published online 21 December 2010.
Berglundh, T., Liljenberg, B., Tarkowski, A. & Lindhe, J. The presence of local and circulating autoreactive B cells in patients with advanced periodontitis. J. Clin. Periodontol. 29, 281–286 (2002).
Jagannathan, M. et al. TLR cross-talk specifically regulates cytokine production by B cells from chronic inflammatory disease patients. J. Immunol. 83, 7461–7470 (2009).
McInnes, I.B. & O'Dell, J.R. State-of-the-art: rheumatoid arthritis. Ann. Rheum. Dis. 69, 1898–1906 (2010).
Bax, M., van Heemst, J., Huizinga, T.W. & Toes, R.E. Genetics of rheumatoid arthritis: what have we learned? Immunogenetics 63, 459–466 (2011).
Katz, J., Goultschin, J., Benoliel, R. & Brautbar, C. Human leukocyte antigen (HLA) DR4. Positive association with rapidly progressing periodontitis. J. Periodontol. 58, 607–610 (1987).
Stein, J., Reichert, S., Gautsch, A. & Machulla, H.K.G. Are there HLA combinations typical supporting for or making resistant against aggressive andsol;or chronic periodontitis? J. Periodontal. Res. 38, 508–517 (2003).
Dyer, J.K. et al. HLA-D types and serum IgG responses to Capnocytophaga in diabetes and periodontitis. J. Dent. Res. 76, 1825–1832 (1997).
Alley, C.S. et al. HLA-D and T lymphocyte reactivity to specific periodontal pathogens in type 1 diabetic periodontitis. J. Periodontol. 64, 974–979 (1993).
Machulla, H.K., Stein, J., Gautsch, A., Langner, J., Schaller, H.G. & Reichert, S. HLA-A, B, Cw, DRB1, DRB3sol;4sol;5, DQB1 in German patients suffering from rapidly progressive periodontitis (RPP) and adult periodontitis (AP). J. Clin. Periodontol. 29, 573–579 (2002).
Bonfil, J.J. et al. A “case control” study on the role of HLA DR4 in severe periodontitis and rapidly progressive periodontitis—identification of types and subtypes using molecular biology (PCR.SSO). J. Clin. Periodontol. 26, 77–84 (1999).
Reinholdt, J., Bay, I. & Svejgaard, A. Association between HLA-antigens and periodontal disease. J. Dent. Res. 56, 1261–1263 (1977).
Moses, J.H., Tsichti, H., Donaldson, P., Smith, P.B., Johnson, N.W. & Bodmer, J.G. HLA and susceptibility to juvenile periodontitis in Afro-Caribbeans. Tissue Antigens 43, 316–319 (1994).
Takashiba, S., Ohyama, H., Oyaizu, K., Kogoe-Kato, N. & Murayama, Y. HLA genetics for diagnosis of susceptibility to early-onset periodontitis. J. Periodontal. Res. 34, 374–378 (1999).
Stein, J.M., Machulla, H.K., Smeets, R., Lampert, F. & Reichert, S. Human leukocyte antigen polymorphism in chronic and aggressive periodontitis among Caucasians: a meta-analysis. J. Clin. Periodontol. 35, 183–192 (2008).
Calsina, G., Ramon, J.M. & Echeverria, J.J. Effects of smoking on periodontal tissues. J. Clin. Periodontol. 29, 771–776 (2002).
Ah, M.K., Johnson, G.K., Kaldahl, W.B., Patil, K.D. & Kalkwarf, K.L. The effect of smoking on the response to periodontal therapy. J. Clin. Periodontol. 21, 91–97 (1994).
Haffajee, A.D. & Socransky, S.S. Relationship of cigarette smoking to the subgingival microbiota. J. Clin. Periodontol. 28, 377–388 (2001).
Zambon, J.J. Periodontal diseases: microbial factors. Ann. Periodontol. 1, 879–925 (1996).
Palmer, R.M., Wilson, R.F., Hasan, A.S. & Scott, D.A. Mechanisms of action of environmental factors—tobacco smoking. J. Clin. Periodontol. 32, 180–195 (2005).
Klareskog, L., Malmstrom, V., Lundberg, K., Padyukov, L. & Alfredsson, L. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin. Immunol. 23, 92–98 (2011).
Klareskog, L. et al. A new model for an etiology of rheumatoid arthritis. Arthritis Rheum. 54, 38–46 (2006).
Makrygiannakis, D. et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann. Rheum. Dis. 67, 1488–1492 (2008).
Padyukov, L., Silva, C., Stolt, P., Alfredsson, L., Klareskog, L. & Epidemiological Invest R A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 50, 3085–3092 (2004).
Linn-Rasker, S.P. et al. Smoking is a risk factor for anti-CCP antibodies only in rheumatoid arthritis patients who carry HLA-DRB1 shared epitope alleles. Ann. Rheum. Dis. 65, 366–371 (2006).
Pedersen, M. et al. Strong combined gene-environment effects in anti-cyclic citrullinated peptide-positive rheumatoid arthritis—a nationwide case-control study in Denmark. Arthritis Rheum. 56, 1446–1453 (2007).
Lundstrom, E., Kallberg, H., Alfredsson, L., Klareskog, L. & Padyukov, L. Gene-environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheum. 60, 1597–1603 (2009).
Hutchinson, D., Shepstone, L., Moots, R., Lear, J.T. & Lynch, M.P. Heavy cigarette smoking is strongly associated with rheumatoid arthritis (RA), particularly in patients without a family history of RA. Ann. Rheum. Dis. 60, 223–227 (2001).
Kallberg, H. et al. Smoking is a major preventable risk factor for rheumatoid arthritis: estimations of risks after various exposures to cigarette smoke. Ann. Rheum. Dis. 70, 508–511 (2011).
Karlson, E.W., Lee, I.M., Cook, N.R., Manson, J.E., Buring, J.E. & Hennekens, C.H. A retrospective cohort study of cigarette smoking and risk of rheumatoid arthritis in female health professionals. Arthritis Rheum. 42, 910–917 (1999).
Mikuls, T.R. et al. Antibody responses to Porphyromonas gingivalis in subjects with rheumatoid arthritis and periodontitis. Int. Immunopharmacol. 9, 38–42 (2009).
Finckh, A., Dehler, S., Costenbader, K.H., Gabay, C. & SCQM. Cigarette smoking and radiographic progression in rheumatoid arthritis. Ann. Rheum. Dis. 66, 1066–1071 (2007).
Manfredsdottir, V.F. et al. The effects of tobacco smoking and rheumatoid factor seropositivity on disease activity and joint damage in early rheumatoid arthritis. Rheumatology 45, 734–740 (2006).
Papadopoulos, N.G., Alamanos, Y., Voulgarl, P.V., Epagelis, E.K., Tsifetaki, N. & Drosos, A.A. Does cigarette smoking influence disease expression, activity and severity in early rheumatoid arthritis patients?. Clin. Exp. Rheumatol. 23, 861–866 (2005).
Lee, H.S. et al. Interaction between smoking, the, shared epitope, and anti-cyclic citrullinated peptide—a mixed picture in three large North American rheumatoid arthritis cohorts. Arthritis Rheum. 56, 1745–1753 (2007).
Haffajee, A.D., Bogren, A., Hasturk, H., Feres, M., Lopez, N.J. & Socransky, S.S. Subgingival microbiota of chronic periodontitis subjects from different geographic locations. J. Clin. Periodontol. 31, 996–1002 (2004).
Masson-Bessiere, C. et al. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J. Immunol. 166, 4177–4184 (2001).
Vossenaar, E.R. et al. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res. Ther. 6, R142–R150 (2004).
Burkhardt, H., Sehnert, B., Bockermann, R., Engstrom, A., Kalden, J.R. & Holmdahl, R. Humoral immune response to citrullinated collagen type II determinants in early rheumatoid arthritis. Eur. J. Immunol. 35, 1643–1652 (2005).
Lundberg, K. et al. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res. Ther. 7, R458–R467 (2005).
Acknowledgements
We thank Professor IB McInnes and Mr Lee Savarrio for kindly critically reviewing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
JPB and SC receive funding from EUFP7 “Gums and Joints” Grant number 261460.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Farquharson, D., Butcher, J. & Culshaw, S. Periodontitis, Porphyromonas, and the pathogenesis of rheumatoid arthritis. Mucosal Immunol 5, 112–120 (2012). https://doi.org/10.1038/mi.2011.66
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2011.66
This article is cited by
-
The Oral-Vascular-Pulmonary Infection Route: a Pathogenic Mechanism Linking Oral Health Status to Acute and Post-Acute COVID-19
Current Oral Health Reports (2023)
-
Interplay of Microbiota and Citrullination in the Immunopathogenesis of Rheumatoid Arthritis
Probiotics and Antimicrobial Proteins (2022)
-
Troublesome friends within us: the role of gut microbiota on rheumatoid arthritis etiopathogenesis and its clinical and therapeutic relevance
Clinical and Experimental Medicine (2021)
-
Gums and joints: is there a connection? Part two: the biological link
British Dental Journal (2019)
-
An interplay of structure and intrinsic disorder in the functionality of peptidylarginine deiminases, a family of key autoimmunity-related enzymes
Cellular and Molecular Life Sciences (2019)
