
Abstract
Previous experience in the treatment of chronic myeloid leukaemia (CML) has shown that the achievement of clinical, morphological and cytogenetic remission does not indicate eradication of the disease. A complete molecular response (CMR; no detectable BCR–ABL mRNA) represents a deeper level of response, but even CMR is not a guarantee of elimination of the leukaemia, because the significance of CMR is determined by the detection limit of the assay that is used. Two studies of imatinib cessation in CMR are underway, cumulatively involving over 100 patients. The current estimated rate of stable CMR after stopping imatinib is approximately 40%, but the duration of follow-up is relatively short. The factors that determine relapse risk are yet to be identified. The intrinsic capacity of any residual leukaemic cells to proliferate following the withdrawal of treatment may be important, but there may also be a role for immunological suppression of the leukaemic clone. No currently available test can formally prove that the leukaemic clone is eradicated. Here we discuss the sensitive measurement of minimal residual disease, and speculate on the biology of BCR–ABL-positive cells that may persist after effective therapy of CML.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
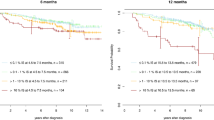

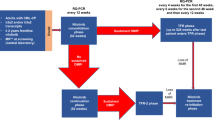
Change history
12 November 2010
This article has been corrected since Advance Online Publication and an erratum is also printed in this issue
References
Mauro MJ, Druker BJ, Maziarz RT . Divergent clinical outcome in two CML patients who discontinued imatinib therapy after achieving a molecular remission. Leuk Res 2004; 28 (Suppl 1): S71–S73.
Cortes J, O'Brien S, Kantarjian H . Discontinuation of imatinib therapy after achieving a molecular response. Blood 2004; 104: 2204–2205.
Branford S, Cross NC, Hochhaus A, Radich J, Saglio G, Kaeda J et al. Rationale for the recommendations for harmonizing current methodology for detecting BCR-ABL transcripts in patients with chronic myeloid leukaemia. Leukemia 2006; 20: 1925–1930.
Branford S, Fletcher L, Cross NC, Muller MC, Hochhaus A, Kim DW et al. Desirable performance characteristics for BCR-ABL measurement on an international reporting scale to allow consistent interpretation of individual patient response comparison of response rates between clinical trials. Blood 2008; 112: 3330–3338.
Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review recommendations for harmonizing current methodology for detecting BCR-ABL transcripts kinase domain mutations for expressing results. Blood 2006; 108: 28–37.
O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F et al. Imatinib compared with interferon low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003; 348: 994–1004.
Guilhot F, Chastang C, Michallet M, Guerci A, Harousseau JL, Maloisel F et al. Interferon alfa-2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. French Chronic Myeloid Leukemia Study Group. N Engl J Med 1997; 337: 223–229.
Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med 2003; 349: 1423–1432.
Branford S, Seymour JF, Grigg A, Arthur C, Rudzki Z, Lynch K et al. BCR-ABL messenger RNA levels continue to decline in patients with chronic phase chronic myeloid leukemia treated with imatinib for more than 5 years approximately half of all first-line treated patients have stable undetectable BCR-ABL using strict sensitivity criteria. Clin Cancer Res 2007; 13: 7080–7085.
Muller MC, Gattermann N, Lahaye T, Deininger MW, Berndt A, Fruehauf S et al. Dynamics of BCR-ABL mRNA expression in first-line therapy of chronic myelogenous leukemia patients with imatinib or interferon alpha/ara-C. Leukemia 2003; 17: 2392–2400.
Ross DM, Watkins DB, Hughes TP, Branford S . Reverse transcription with random pentadecamer primers improves the detection limit of a quantitative PCR assay for BCR-ABL transcripts in chronic myeloid leukemia: implications for defining sensitivity in minimal residual disease. Clin Chem 2008; 54: 1568–1571.
Cross NC, Hughes TP, Feng L, O'Shea P, Bungey J, Marks DI et al. Minimal residual disease after allogeneic bone marrow transplantation for chronic myeloid leukaemia in first chronic phase: correlations with acute graft-versus-host disease relapse. Br J Haematol 1993; 84: 67–74.
Melo JV, Yan XH, Diamond J, Lin F, Cross NC, Goldman JM . Reverse transcription/polymerase chain reaction (RT/PCR) amplification of very small numbers of transcripts: the risk in misinterpreting negative results. Leukemia 1996; 10: 1217–1221.
Ross DM, Branford S, Melo JV, Hughes TP . Reply to ‘What do we mean by sensitivity when we talk about detecting minimal residual disease?’ by Steinbach and Debatin. Leukemia 2009; 23: 819–820;author reply 820.
Bose S, Deininger M, Gora-Tybor J, Goldman JM, Melo JV . The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance implications for the assessment of minimal residual disease. Blood 1998; 92: 3362–3367.
Biernaux C, Loos M, Sels A, Huez G, Stryckmans P . Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood 1995; 86: 3118–3122.
Mattarucchi E, Spinelli O, Rambaldi A, Pasquali F, Lo Curto F, Campiotti L et al. Molecular monitoring of residual disease in chronic myeloid leukemia by genomic DNA compared with conventional mRNA analysis. J Mol Diagn 2009; 11: 482–487.
Sobrinho-Simoes M, Wilczek V, Score J, Cross NC, Apperley JF, Melo JV . In search of the original leukemic clone in chronic myeloid leukemia patients in complete molecular remission after stem cell transplantation or imatinib. Blood 2010; 116: 1329–1335.
Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Bartley PA et al. Patients with chronic myeloid leukaemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukaemia by DNA PCR. Leukemia 2010; in press.
Bartley PA, Ross DM, Latham S, Martin-Harris MH, Budgen B, Wilczek V et al. Sensitive detection quantification of minimal residual disease in chronic myeloid leukaemia using nested quantitative PCR for BCR-ABL DNA. Int J Lab Hematol 2010; e-pub ahead of print 10 May 2010.
van der Velden VH, Panzer-Grumayer ER, Cazzaniga G, Flohr T, Sutton R, Schrauder A et al. Optimization of PCR-based minimal residual disease diagnostics for childhood acute lymphoblastic leukemia in a multi-center setting. Leukemia 2007; 21: 706–713.
Rawer D, Borkhardt A, Wilda M, Kropf S, Kreuder J . Influence of stochastics on quantitative PCR in the detection of minimal residual disease. Leukemia 2003; 17: 2527–2528; author reply 2528–2531.
Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL et al. Dynamics of chronic myeloid leukaemia. Nature 2005; 435: 1267–1270.
Roeder I, Horn M, Glauche I, Hochhaus A, Mueller MC, Loeffler M . Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights clinical implications. Nat Med 2006; 12: 1181–1184.
Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 2004; 351: 657–667.
Forrest DL, Trainor S, Brinkman RR, Barnett MJ, Hogge DE, Nevill TJ et al. Cytogenetic molecular responses to standard-dose imatinib in chronic myeloid leukemia are correlated with Sokal risk scores duration of therapy but not trough imatinib plasma levels. Leuk Res 2009; 33: 271–275.
Larson RA, Druker BJ, Guilhot F, O'Brien SG, Riviere GJ, Krahnke T et al. Imatinib pharmacokinetics and its correlation with response safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood 2008; 111: 4022–4028.
Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia 2007; 21: 926–935.
Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L et al. Primitive quiescent Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002; 99: 319–325.
Dorsey JF, Cunnick JM, Lanehart R, Huang M, Kraker AJ, Bhalla KN et al. Interleukin-3 protects Bcr-Abl-transformed hematopoietic progenitor cells from apoptosis induced by Bcr-Abl tyrosine kinase inhibitors. Leukemia 2002; 16: 1589–1595.
Holyoake TL, Jiang X, Jorgensen HG, Graham S, Alcorn MJ, Laird C et al. Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3. Blood 2001; 97: 720–728.
Hirano N, Takahashi T, Ohtake S, Hirashima K, Emi N, Saito K et al. Expression of costimulatory molecules in human leukemias. Leukemia 1996; 10: 1168–1176.
Yong AS, Szydlo RM, Goldman JM, Apperley JF, Melo JV . Molecular profiling of CD34+ cells identifies low expression of CD7 along with high expression of proteinase 3 or elastase as predictors of longer survival in patients with CML. Blood 2006; 107: 205–212.
Holyoake T, Jiang X, Eaves C, Eaves A . Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 1999; 94: 2056–2064.
Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 2006; 107: 4532–4539.
Lenaerts T, Pacheco JM, Traulsen A, Dingli D . Tyrosine kinase inhibitor therapy can cure chronic myeloid leukemia without hitting leukemic stem cells. Haematologica 2010; 95: 900–907.
Goldman J, Gordon M . Why do chronic myelogenous leukemia stem cells survive allogeneic stem cell transplantation or imatinib: does it really matter? Leuk Lymphoma 2006; 47: 1–7.
Clift RA, Appelbaum FR, Thomas ED . Treatment of chronic myeloid leukemia by marrow transplantation. Blood 1993; 82: 1954–1956.
Kaeda J, O'Shea D, Szydlo RM, Olavarria E, Dazzi F, Marin D et al. Serial measurement of BCR-ABL transcripts in the peripheral blood after allogeneic stem cell transplantation for chronic myeloid leukemia: an attempt to define patients who may not require further therapy. Blood 2006; 107: 4171–4176.
Radich JP, Gehly G, Gooley T, Bryant E, Clift RA, Collins S et al. Polymerase chain reaction detection of the BCR-ABL fusion transcript after allogeneic marrow transplantation for chronic myeloid leukemia: results and implications in 346 patients. Blood 1995; 85: 2632–2638.
Talpaz M, McCredie KB, Mavligit GM, Gutterman JU . Leukocyte interferon-induced myeloid cytoreduction in chronic myelogenous leukemia. Blood 1983; 62: 689–692.
Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009; 458: 904–908.
Selleri C, Sato T, Del Vecchio L, Luciano L, Barrett AJ, Rotoli B et al. Involvement of Fas-mediated apoptosis in the inhibitory effects of interferon-alpha in chronic myelogenous leukemia. Blood 1997; 89: 957–964.
Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE et al. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med 2000; 6: 1018–1023.
Burchert A, Wolfl S, Schmidt M, Brendel C, Denecke B, Cai D et al. Interferon-alpha, but not the ABL-kinase inhibitor imatinib (STI571), induces expression of myeloblastin and a specific T-cell response in chronic myeloid leukemia. Blood 2003; 101: 259–264.
Molldrem JJ, Lee PP, Kant S, Wieder E, Jiang W, Lu S et al. Chronic myelogenous leukemia shapes host immunity by selective deletion of high-avidity leukemia-specific T cells. J Clin Invest 2003; 111: 639–647.
Guilhot F, Roy L, Guilhot J, Millot F . Interferon therapy in chronic myelogenous leukemia. Hematol Oncol Clin North Am 2004; 18: 585–603, viii.
Mahon FX, Delbrel X, Cony-Makhoul P, Faberes C, Boiron JM, Barthe C et al. Follow-up of complete cytogenetic remission in patients with chronic myeloid leukemia after cessation of interferon alfa. J Clin Oncol 2002; 20: 214–220.
Verbeek W, Konig H, Boehm J, Kohl D, Lange C, Heuer T et al. Continuous complete hematological and cytogenetic remission with molecular minimal residual disease 9 years after discontinuation of interferon-alpha in a patient with Philadelphia chromosome-positive chronic myeloid leukemia. Acta Haematol 2006; 115: 109–112.
Bonifazi F, de Vivo A, Rosti G, Guilhot F, Guilhot J, Trabacchi E et al. Chronic myeloid leukemia and interferon-alpha: a study of complete cytogenetic responders. Blood 2001; 98: 3074–3081.
Kantarjian HM, O'Brien S, Cortes JE, Shan J, Giles FJ, Rios MB et al. Complete cytogenetic and molecular responses to interferon-alpha-based therapy for chronic myelogenous leukemia are associated with excellent long-term prognosis. Cancer 2003; 97: 1033–1041.
Burchert A, Muller MC, Kostrewa P, Erben P, Bostel T, Liebler S et al. Sustained molecular response with interferon alfa maintenance after induction therapy with imatinib plus interferon alfa in patients with chronic myeloid leukemia. J Clin Oncol 2010; 28: 1429–1435.
Rousselot P, Huguet F, Rea D, Legros L, Cayuela JM, Maarek O et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood 2007; 109: 58–60.
Verma D, Kantarjian H, Jain N, Cortes J . Sustained complete molecular response after imatinib discontinuation in a patient with chronic myeloid leukemia not previously exposed to interferon alpha. Leuk Lymphoma 2008; 49: 353–1402.
Mahon FX, Rea D, Guilhot F, Huguet F, Nicolini FE, Legros L et al. Discontinuation of imatinib therapy after achieving a complete molecular response in chronic myeloid leukemia patients. Blood 2009; 114: 353 (abstract no. 859).
Chen CI, Maecker HT, Lee PP . Development and dynamics of robust T-cell responses to CML under imatinib treatment. Blood 2008; 111: 5342–5349.
Rezvani K, Grube M, Brenchley JM, Sconocchia G, Fujiwara H, Price DA et al. Functional leukemia-associated antigen-specific memory CD8+ T cells exist in healthy individuals and in patients with chronic myelogenous leukemia before and after stem cell transplantation. Blood 2003; 102: 2892–2900.
Barrett AJ, Rezvani K . Translational mini-review series on vaccines: Peptide vaccines for myeloid leukaemias. Clin Exp Immunol 2007; 148: 189–198.
Pinilla-Ibarz J, Cathcart K, Korontsvit T, Soignet S, Bocchia M, Caggiano J et al. Vaccination of patients with chronic myelogenous leukemia with bcr-abl oncogene breakpoint fusion peptides generates specific immune responses. Blood 2000; 95: 1781–1787.
Rojas JM, Knight K, Wang L, Clark RE . Clinical evaluation of BCR-ABL peptide immunisation in chronic myeloid leukaemia: results of the EPIC study. Leukemia 2007; 21: 2287–2295.
Bocchia M, Gentili S, Abruzzese E, Fanelli A, Iuliano F, Tabilio A et al. Effect of a p210 multipeptide vaccine associated with imatinib or interferon in patients with chronic myeloid leukaemia and persistent residual disease: a multicentre observational trial. Lancet 2005; 365: 657–662.
DeAngelo DJ, Hochberg EP, Alyea EP, Longtine J, Lee S, Galinsky I et al. Extended follow-up of patients treated with imatinib mesylate (gleevec) for chronic myelogenous leukemia relapse after allogeneic transplantation: durable cytogenetic remission and conversion to complete donor chimerism without graft-versus-host disease. Clin Cancer Res 2004; 10: 5065–5071.
Savani BN, Montero A, Kurlander R, Childs R, Hensel N, Barrett AJ . Imatinib synergizes with donor lymphocyte infusions to achieve rapid molecular remission of CML relapsing after allogeneic stem cell transplantation. Bone Marrow Transplant 2005; 36: 1009–1015.
Butt NM, Rojas JM, Wang L, Christmas SE, Abu-Eisha HM, Clark RE . Circulating bcr-abl-specific CD8+ T cells in chronic myeloid leukemia patients and healthy subjects. Haematologica 2005; 90: 1315–1323.
Bustamante JM, Bixby LM, Tarleton RL . Drug-induced cure drives conversion to a stable and protective CD8+ T central memory response in chronic Chagas disease. Nat Med 2008; 14: 542–550.
Mumprecht S, Schurch C, Schwaller J, Solenthaler M, Ochsenbein AF . Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood 2009; 114: 1528–1536.
Dietz AB, Souan L, Knutson GJ, Bulur PA, Litzow MR, Vuk-Pavlovic S . Imatinib mesylate inhibits T-cell proliferation in vitro and delayed-type hypersensitivity in vivo. Blood 2004; 104: 1094–1099.
Schade AE, Schieven GL, Townsend R, Jankowska AM, Susulic V, Zhang R et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood 2008; 111: 1366–1377.
Blake S, Hughes TP, Mayrhofer G, Lyons AB . The Src/ABL kinase inhibitor dasatinib (BMS-354825) inhibits function of normal human T-lymphocytes in vitro. Clin Immunol 2008; 127: 330–339.
Blake SJ, Lyons AB, Fraser CK, Hayball JD, Hughes TP . Dasatinib suppresses in vitro natural killer cell cytotoxicity. Blood 2008; 111: 4415–4416.
Blake SJ, Lyons AB, Hughes TP . Nilotinib inhibits the Src-family kinase LCK and T-cell function in vitro. J Cell Mol Med 2009; 13: 599–601.
Mustjoki S, Ekblom M, Arstila TP, Dybedal I, Epling-Burnette PK, Guilhot F et al. Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia 2009; 23: 1398–1405.
Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood 2002; 99: 3530–3539.
Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT et al. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood 2002; 100: 1965–1971.
Melo JV, Barnes DJ . Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer 2007; 7: 441–453.
Koptyra M, Falinski R, Nowicki MO, Stoklosa T, Majsterek I, Nieborowska-Skorska M et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood 2006; 108: 319–327.
Nowicki MO, Falinski R, Koptyra M, Slupianek A, Stoklosa T, Gloc E et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood 2004; 104: 3746–3753.
Sallmyr A, Tomkinson AE, Rassool FV . Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood 2008; 112: 1413–1423.
Dierov J, Sanchez PV, Burke BA, Padilla-Nash H, Putt ME, Ried T et al. BCR/ABL induces chromosomal instability after genotoxic stress and alters the cell death threshold. Leukemia 2009; 23: 279–286.
Breedveld P, Beijnen JH, Schellens JH . Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol Sci 2006; 27: 17–24.
Peng B, Lloyd P, Schran H . Clinical pharmacokinetics of imatinib. Clin Pharmacokinet 2005; 44: 879–894.
White DL, Saunders VA, Dang P, Engler J, Venables A, Zrim S et al. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood 2007; 110: 4064–4072.
Burger H, van Tol H, Boersma AW, Brok M, Wiemer EA, Stoter G et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004; 104: 2940–2942.
Thomas J, Wang L, Clark RE, Pirmohamed M . Active transport of imatinib into and out of cells: implications for drug resistance. Blood 2004; 104: 3739–3745.
Yong AS, Keyvanfar K, Hensel N, Eniafe R, Savani BN, Berg M et al. Primitive quiescent CD34+ cells in chronic myeloid leukemia are targeted by in vitro expanded natural killer cells, which are functionally enhanced by bortezomib. Blood 2009; 113: 875–882.
Dillmann F, Veldwijk MR, Laufs S, Sperandio M, Calandra G, Wenz F et al. Plerixafor inhibits chemotaxis toward SDF-1 and CXCR4-mediated stroma contact in a dose-dependent manner resulting in increased susceptibility of BCR-ABL(+) cell to Imatinib and Nilotinib. Leuk Lymphoma 2009; 55: 1676–1686.
Jin L, Tabe Y, Konoplev S, Xu Y, Leysath CE, Lu H et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol Cancer Ther 2008; 7: 48–58.
Barnes DJ, Schultheis B, Adedeji S, Melo JV . Dose-dependent effects of Bcr-Abl in cell line models of different stages of chronic myeloid leukemia. Oncogene 2005; 24: 6432–6440.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Ross, D., Hughes, T. & Melo, J. Do we have to kill the last CML cell?. Leukemia 25, 193–200 (2011). https://doi.org/10.1038/leu.2010.197
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2010.197
Keywords
This article is cited by
-
Elevated plasma levels of IL-6 and MCP-1 selectively identify CML patients who better sustain molecular remission after TKI withdrawal
Journal of Hematology & Oncology (2023)
-
Discontinuation of tyrosine kinase inhibitor in chronic myeloid leukemia: a retrospective cohort in east occitania
Annals of Hematology (2022)
-
Lineage of measurable residual disease in patients with chronic myeloid leukemia in treatment-free remission
Leukemia (2020)
-
Treatment-free remission in patients with chronic myeloid leukaemia
Nature Reviews Clinical Oncology (2020)
-
Chronic myeloid leukemia stem cells and molecular target therapies for overcoming resistance and disease persistence
International Journal of Hematology (2018)
