
Abstract
Collagen type III glomerulopathy (Col3GP) is a rare renal disease characterized by massive glomerular accumulations of collagen type III. The disease occurs in both humans and animals, and has been presumed to be heritable with an autosomal recessive inheritance pattern. The pathogenesis is unknown. We describe herein a condition of canine autosomal recessive Col3GP. This spontaneously occurring canine disease was incidentally diagnosed in six mongrel dogs. We then established and studied a pedigree segregating the disease to confirm the genetic nature and inheritance of canine Col3GP. Twenty-nine percent of offspring (14/48) were affected, strongly supporting a simple autosomal recessive inheritance pattern. Kidney specimens were studied by light microscopy, electron microscopy (EM), immunohistochemistry and in situ hybridization. Characteristic findings of Col3GP previously reported in both humans and animals were demonstrated, including massive glomerular collagen type III deposition, and evidence of local mesangial collagen type III synthesis was found. We propose that canine Col3GP may serve as an animal model of human Col3GP. Our initial studies, using simple segregation analysis, showed that the Col3A1 gene was not involved in the disease. This is the first animal model of Col3GP, and further studies of this phenotype in dogs may have the potential to provide information on the pathogenesis and genetics of the disease in both animals and humans, and may thus contribute to the development of treatment regimes.
Similar content being viewed by others

Main
Collagen type III glomerulopathy (Col3GP), also known as collagenofibrotic glomerulopathy, is a rare non-immunoglobulin derived glomerular disease with organized extracellular deposits. It is characterized by extensive accumulations of collagen type III in the capillary walls and the mesangial matrix of the glomeruli. The cross-striated curved and frayed collagen fibrils can be visualized by EM and detected by immunohistochemistry. About 50 human cases have been reported in the English literature, with the majority of the patients from Japan.1 Col3GP has no sex predilection and can affect both children and adults. The typical clinical features are proteinuria, hypertension and a variable degree of slowly progressive renal impairment, and the disease tends to have a more aggressive course in children than in adults.2 In veterinary medicine Col3GP is reported to occur in various animal species, including monkeys,3, 4 dogs,5, 6, 7 pigs8 and a cat.9 The pathogenic mechanisms of Col3GP are obscure, and it is not clarified whether it is a primary renal disease in which the collagen is produced in the glomeruli itself or a systemic disease where the collagen or its precursor is derived from an extrarenal source.1 In both animals7 and humans10, 11 Col3GP has been reported in siblings of healthy parents, and this has led to the assumption that Col3GP is a heritable disease with an autosomal recessive inheritance. The aim of this study was to provide a precise pathoanatomical description of the Col3GP phenotype in the studied dogs and to elucidate possible sources of the excess collagen type III. Furthermore we aimed to establish an inheritance pattern of this canine variant of the disease, and specifically check if the Col3A1 gene is associated with the disorder. This naturally occurring canine disease is the first animal model of human Col3GP.
MATERIALS AND METHODS
Animals
Col3GP was incidentally diagnosed in six mongrel puppies born in a family segregating canine hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis (RCND), a hereditary renal cancer syndrome in dogs,12, 13 at the Norwegian School of Veterinary Science. Five litters produced by a male and two female carriers of Col3GP were studied. In total 35 puppies were born, eight of which were affected by Col3GP. The age of the 14 Col3GP-positive dogs at euthanasia ranged from 41 to 326 days. Blood analysis including serum biochemistry and hematology was available in eight cases, in three cases only serum biochemistry was available and in three cases blood analysis was lacking. Blood samples were collected before euthanasia. Urinary protein:creatinin ratio performed on bladder urine at autopsy was available in four cases. The study was approved and performed in accordance with the National animal welfare rules and regulation for research involving animals (LOV-2009-06-19-97/FOR-1996-01-15-23).
Preparation of Renal Tissue
Autopsy specimens from the kidneys of the affected dogs were studied by light microscopy (LM) (14 specimens), EM (10 specimens), immunohistochemical analysis (4 specimens) and in situ hybridization (2 specimens). Renal tissues from healthy siblings of the same age were used as controls. At autopsy the kidneys were perfused with 0.9% saline. Slices of renal tissues were fast frozen in Freon cooled in liquid nitrogen and stored at −80 °C for immunohistochemical, immunofluorescence and mRNA in situ hybridization studies.
Light microscopy
Slices of renal tissues that included both medulla and cortex were fixed by immersion in 10% buffered formaldehyde and embedded in paraffin. Two to four micrometer thick sections were stained with hematoxylin and eosin, periodic acid-schiff (PAS), van Gieson, Masson’s trichrome and Congo red and studied under a Nikon Eclipse 50i light microscope. Photomicrographs were captured by a Nikon DS-Fi1 camera.
Immunohistochemistry and immunofluorescence
The tissues were stained for collagen type III, fibronectin and smooth muscle actin (SMA) using polymer-based immunohistochemistry, and for C3 and immunoglobulins using indirect immunofluorescence. For immunohistochemistry the primary antibodies used were monoclonal mouse anti-human SMA (α-SMA) (DAKO, Glostrup, Denmark), polyclonal rabbit anti-human fibronectin (DAKO), mouse anti-monoclonal antibody to collagen type III (Acris Antibodies, Herford, Germany), mouse anti-collagen III antibody (Novus Biologicals, Littleton, CO, USA). Secondary antibodies used were anti-mouse and anti-rabbit IgG conjugated with a horseradish peroxidase (HRP) labeled polymer, from the EnVision+ System-HRP kit (DAKO). The sections were treated with a diaminobenzidine hydrochloric acid solution from the EnVision+ kit, and counterstained with hematoxylin. For immunofluorescence goat anti-dog C3 Antibody (Bethyl, Montgomery, TX, USA), goat anti-dog IgM (Bethyl) and goat anti-dog IgG1 (Bethyl) were used as primary antibodies and FITC-conjugated goat IgG Antibody (Bethyl) as secondary antibody. Washing between each of the steps in the procedures was performed with tris-buffered saline or distilled water. The primary antibodies were diluted in a 1% solution of bovine serum albumin in tris-buffered saline. Negative control staining was performed by replacing the primary antibodies with non-immunized goat serum, and showed no staining. Normal dog renal specimens from healthy siblings were used as control. The specimens were studied in a Nikon Eclipse 50i microscope equipped with a Nikon Intensilight C-HGFI fluorescence light source and a DAPI fluorescence filter, and photomicrographs were captured with a Nikon DS-U1 camera.
In situ hybridization
Frozen kidney sections (12 μm) from two affected and one healthy dog were cut with a cryostat (Leitz Cryostat 1720, Germany) and mounted on positively charged slides (Superfrost Plus; Menzel-Gläser; Germany). The sections were air-dried and stored at −80 °C until use. Sense and antisense digoxigenin labeled cRNA probes covering a 300 bp fragment of the coding regions of COLIII mRNA were prepared following the manufacturer’s instructions (Roche, Germany). In situ hybridization was carried out according to the method of Barthel and Raymond14 with some modifications. Briefly, the tissue sections were rehydrated, fixed in 4% paraformaldehyde in PBS, treated with 10 μg/ml proteinase K (Sigma-Aldrich, St Louis, MO, USA) for 5 min at 37 °C followed by fixation in 4% paraformaldehyde in PBS, then treated with acetic anhydride and dehydrated. Approximately 100 μl hybridization mixture containing 100 ng of each digoxigenin labeled cRNA probe was applied directly to each air-dried section and the sections were incubated in a humidity chamber with coverslips for 15 h at 60 °C. Non-specific binding of probes was removed by digestion for 30 min at 37 °C with 20 μg/ml RNase A (Sigma-Aldrich). The probes were detected by using an anti-digoxigenin antibody coupled to alkaline phosphatase. Labeling was visualized using nitro-blue tetrazolium chloride/5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt as substrate. For each of the analyzed tissue sections, sense probes were applied to serial sections as a negative control to confirm the specificity of the hybridization.
Segregation analysis
Based on the sequence (CanFam 2.1/Ensembl) embracing the Col3A1 gene we identified and designed primers for two (tetranucleotide) microsatellite markers (M1 and M2) in the region. M1 is localized ∼18 kb upstreams of exon1, and M2 is in intron 23 in the gene. Haplotypes containing most of the Col3A1 gene could therefore easily be defined. PCR was performed using fluorescein tagged forward primers for each of the microsatellites (annealing T: 57 °C/30 cycles). The primers used were; M1-F: 5′-AGTCCCATTGCTTTCCACAC-3′ M1-R: T 5′-TTTTCCGTTGTCATTTTGAT-3′ M2-F: 5′-CCGTGTCTCTGCCTCTCTCT-3′ M2-R: 5′-GGACCTGGATGTCCACTTGT-3′ and genotyping was based on simple AFLP using capillary electrophoresis (Applied 3130XL).
Transmission EM
Small pieces of renal tissues were prefixed in 3% glutaraldehyde in 0.1 M cacodylatebuffer, post-fixed in osmium, dehydrated in ethanol and embedded in epon. Semithin sections were stained with toluidine blue and examined under the light microscope for selection of glomeruli. Ultrathin sections were stained with uranyl acetate and lead citrate. The specimens were observed in a Philips EM 208 S electron microscope and photographs were captured by a Mega View III camera.
RESULTS
Breeding Experiments for Segregation of the Col3GP Affected Phenotype
Two females and one male dog produced eight offspring of the Col3GP phenotype. In addition six Col3GP affected dogs had previously been incidentally born in the colony. The 14 Col3GP-positive dogs originated from 7 litters comprising a total number of 48 puppies (29% affected), and included 5 males and 9 females. The parents of the affected puppies were healthy and exhibited no signs of renal impairment. The pattern of segregation for the Col3GP phenotype in this colony was consistent with a simple autosomal recessive mode of inheritance (Figure 1). From this total of 14 dogs of the Col3GP phenotype, seven were affected by another hereditary disease, RCND,12, 13 whereas seven did not have RCND. Clinical signs including anorexia and weight loss were observed in four dogs (Nos. 1, 2, 3 and 4) aged, respectively, 50, 190, 261 and 326 days at euthanasia. Analysis of blood and urinary samples from these dogs revealed proteinuria and azotemia (Table 1). The remaining ten dogs (Nos. 5, 6, 7, 8, 9, 10, 11, 12, 13 and 14) aged, respectively, 41, 57, 57, 73, 80, 80, 80, 91, 91 and 208 days at euthanasia did not show signs of disease or discomfort at physical examination, but two of these (Nos. 8 and 11) exhibited proteinuria and high urine protein:creatinine ratio values at the time of autopsy (Table 1). Case Nos. 12 and 13 showed slightly increased blood urea nitrogen and normal serum creatinine.

A pedigree of affected dogs. The pattern of inheritance and the proportion of affected offspring (29%) are consistent with an autosomal recessive mode of inheritance.
Renal Morphology of the Col3GP Phenotype
Macroscopic findings
Dogs that had developed clinical signs of renal failure exhibited pale and firm kidneys with granular surfaces. Except for two dogs with reduced amounts of muscular mass and tissue pallor (Nos. 2 and 4) indicative of anemia, no extrarenal macroscopic lesions was observed. Among the dogs without clinical signs of renal failure the macroscopic lesions ranged from no findings to moderately enlarged, pale and swollen kidneys (Figure 2).

The kidneys of a collagen type III glomerulopathy (Col3GP) affected individual at the left are pale and enlarged compared with the normal kidneys of a healthy sibling.
Light microscopic lesions
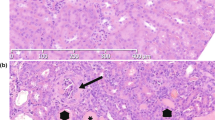
All 14 affected animals revealed essentially identical diffuse glomerular lesions. The mesangium of nearly all glomeruli was severely expanded by a homogeneous eosinophilic material (Figure 3a). The capillary walls were extremely thickened by the same material interposed between the endothelium and the glomerular basement membrane (GBM), and GBM double contours were occasionally observed as a result of interpositioning of this material, thus showing a membranoproliferative appearance, with moderate to severe increased mesangial hypercellularity and focal and segmental endocapillary proliferation. In most glomeruli many capillary lumens were compressed and occasionally effaced. The abnormal extracellular mesangial material was Congo red negative and it stained pale pink in PAS-stained sections (Figure 3b), bright blue in sections stained with Masson’s trichrome (Figure 3c) and bright red in sections stained with Van Gieson (Figure 3d). Some glomeruli were decreased in size within cystically dilated Bowman’s spaces. Glomerular synechiae were occasionally observed. Glomerular obsolescence was frequently observed in individuals where chronic kidney disease had developed (Nos 1, 2, 3 and 4). There was moderate to severe tubular atrophy with occasional distal tubules with luminal proteinaceous casts and a varying degree of periglomerular fibrosis and interstitial fibrosis with expansion of mature collagen. In the mildest affected cases there was no increase in interstitial infiltration of inflammatory cells, whereas multifocal interstitial lymphoplasmacytic and macrophage infiltration occasionally could be observed in the more severely affected individuals. Younger dogs generally exhibited milder tubulointerstitial pathological changes than the older dogs. Arterioles and arteries showed no specific lesions.


Light microscopic appearances of affected glomeruli. (a) Mesangial expansion and thickening of the capillary walls by deposited extracellular eosinophilic fibrillar material, case no. 11 (hematoxylin and eosin, × 40). (b) The deposited material stains weakly positive for periodic acid-schiff, case no. 13 (periodic acid-schiff, × 40). (c, d) The homogenous deposited material stains distinctively blue (Masson’s trichrome, × 40) and red (van Gieson, × 40), suggesting a collagenous nature of the deposited material, case no. 5. (e) The deposits stains intensely positive for collagen type III, case no. 12 (anti-collagen III immunostain, × 20). (f) Increased mesangial and capillary wall staining for fibronectin, case no. 13 (anti-fibronectin immunostain, × 40). (g) Limited staining for α-smooth muscle actin in mesangial cells (anti-α-smooth muscle actin immunostain, × 40), case no. 13 (h) There is weak irregular and patchy-positive reaction for C3 along glomerular capillary walls, case no. 9 (anti-C3 immunofluorescence, × 40). (i) Collagen type III mRNA synthesis is identified in mesangial areas, case no. 7 (collagen type III in situ hybridization, × 40).
Immunohistochemistry and immunofluorescence findings
Immunohistochemical examination revealed that the organized deposits in the mesangial matrix and the capillary walls stained intensely positive for collagen type III (Figure 3e). There was also an increased strong labeling of the mesangial and capillary wall areas for fibronectin (Figure 3f), coinciding with the distribution of the collagen. A few mesangial cells were positive for α-SMA (Figure 3g). Immunofluorescence studies showed weak irregular, sometimes granular staining for C3 along the capillary walls and in mesangial areas corresponding with the collagen accumulations (Figure 3h), and irregular more linear staining for IgM along the capillary walls. Staining for IgG was negative. In comparison, normal glomeruli from control animals stained negative for collagen type III and IgG, positive for fibronectin, showed scarce focal and segmental mesangial staining for α-SMA and C3 and positive mesangial staining for IgM.
In situ hybridization findings
In order to identify collagen type III mRNA synthesis, kidney sections from normal and affected animals were analyzed by in situ hybridization. Normal glomeruli showed no hybridization; whereas distinct increased signals of collagen type III mRNA expression were observed in glomerular mesangial areas of affected animals (Figure 3i). No signals were detected with the sense probe.
Segregation analysis excluding the Col3A1 gene
As the Col3A1 gene was a likely candidate gene for mutations causing the disease, and we had pedigree information/DNA from all family members, we performed a simple segregation analysis involving the gene in 16 informative offspring. The studied parents were heterozygotes for marker haplotypes involving the Col3A1 gene. As both parents also were heterozygote for the disease all affected offspring would need to inherit the same two haplotypes from the parents, and all healthy animals would have a different combination if the gene had been involved. The results showed that all four possible Col3A1 haplotypes were represented in both affected and unaffected offspring, proving that the locus is not associated with mutations in Col3GP.
Transmission electron microscopic lesions
All studied specimens exhibited similar glomerular ultrastructural pathological changes with severe expansion of the mesangial matrix and the subendothelial aspect of the capillary walls by an amorphous lucent material containing darker fibrils (Figure 4a). The fibrils showed a marked variation in diameter and sometimes appeared in bundles, but more often they were detached, frayed and curved. The larger fibrils were cross-striated with a faint periodicity of ∼60 nm (Figure 4b). In various cases the bundles of fibrils were observed close to mesangial cells and often appeared in close relation to protrusions of the mesangial cellular membrane and cytoplasm (Figure 4c). The expansions of the capillary walls by collagen were most widespread at the ends of the capillary walls where the capillary walls verge with the mesangium (Figure 4a). The GBM was in most cases considered to be within normal limits; however, occasionally the lamina densa was slightly thickened and contained an irregular outer surface with protrusions to the subepithelial aspect. The podocytes exhibited a varying degree of foot process effacement, and focal microvillus transformation was frequently observed. Endothelial swelling with loss of fenestration was a frequent finding. There were no electron-dense immune complex type deposits in the capillary wall or mesangium. The interstitium revealed increased amounts of apparently normal, straight interstitial collagen with distinct transverse band structure, well organized in bundles.

Transmission electron microscopy. (a) The mesangium and the subendothelial area are strikingly widened by massive accumulations of deposited collagen fibrils, with normal lamina densa and segmental foot process effacement, case no.13. Bar=2 μm. (b) Bundles of collagen fibrils seemingly protruding from a mesangial cell, case no. 13. Bar=1 μm. (c) Some of the fibrils reveal cross striations with a periodicity of ∼60 nm, case no. 12. Bar=500 nm.
DISCUSSION
Discrepancy concerning the term of this disease exists. ‘Primary glomerular fibrosis’ was used by Ikeda et al15 in 1990 after identifying the fibrils as collagen type III. Since then ‘collagenofibrotic glomerulonephropathy’, ‘collagenofibrotic glomerulopathy’16, 17 and ‘Col3GP’2, 18 have been in use. We have chosen to apply the term Col3GP because it describes the anatomical location of the primary pathology and precisely accounts for the characteristics of the glomerulopathy.
Pedigree analysis demonstrated that both sexes are affected by Col3GP, and the parents of the affected puppies showed no signs of kidney disease. The pattern of segregation for the Col3GP phenotype in this pedigree was consistent with a simple autosomal recessive mode of inheritance. Col3GP occurred spontaneously in this colony of dogs that was carrying RCND. RCND is not associated with primary glomerular pathologic changes. Among the 14 Col3GP dogs in the present study, equal numbers were RCND affected and unaffected, supporting that Col3GP is inherited independently from RCND.
The pathological findings of the hereditary glomerular disease occurring in this colony of dogs are consistent with previous reports of Col3GP and collagenofibrotic glomerulopathy, both in humans1, 2, 11 and in animals.3, 5, 8 By LM, massive accumulation of fibrillar material expanded the mesangium and the capillary walls, obliterating the capillary lumens. The deposited material was Congo red negative; hence the possibility of amyloidosis was excluded. The distinctive periodicity of banded collagen was disclosed at the ultrastructural level, excluding other types of glomerular deposits and confirming a diagnosis of pathological collagen deposition. A diagnosis of Col3GP was obtained by immunohistochemical examination and this was supported by in situ hybridization findings.
Mesangial cell hypercellularity observed by LM was a general feature in most of the affected cases in this study. In human and canine Col3GP case reports mesangial hypercellularity was occasionally observed.5, 15, 17, 19, 20, 21 The variability of the observed mesangial hypercellularity in Col3GP affected cases is also demonstrated in our closely related cases, ranging from moderate to severe. At the ultrastructural level the abnormal glomerular collagen fibrils in our studied cases showed a transverse band structure with a periodicity of ∼60 nm, were abnormally curved and seemed to be haphazardly arranged, coinciding with earlier reports of Col3GP in humans and animals. However, most of the abnormal fibrils showed a faint periodicity compared with the characteristics of the normal collagen in the renal interstitium, while other fibrils showed no periodicity. In comparison, the interstitial collagen in affected cases exhibited apparently normal straight collagen fibrils revealing distinct transverse band structure, well organized in bundles. These observations suggest that the abnormal glomerular collagen differs structurally from normal collagen. Except from a human case report describing fibrils with deviant vague periodicity,16 faint periodicity has not been a feature in previous reports of Col3GP. Diverging or absent cross-striation can result from packing defects of the collagen molecules forming the fibrils, due to altered ionic strength, pH, temperature, phosphate concentration and collagen concentration.22 We speculate that the high concentration of collagen in the glomeruli might have had an effect on the composition of the fibrils. We confirmed that this glomerular collagen was often located in close relation to the mesangial cells, we also found that capillary wall collagen accumulation was more widespread where the capillary walls abut the mesangium. Mesangial cells are capable of producing collagen III.23, 24, 25 Our in situ hybridization analysis confirmed ongoing collagen type III production in mesangial cells. The distribution of the positive mesangial cells was, however, more limited than the distribution of the massive collagen accumulation. To the best of our knowledge this is the first report that documents glomerular de novo collagen type III synthesis by in situ hybridization in Col3GP affected humans or animals. These findings suggest that the collagen origins from the mesangium and expands into the subendothelial space of the capillary walls. So far it has been unclear whether the epithelial or the endothelial glomerular cell might be responsible for the abnormal production of collagen type III,1 but based on these morphologic and in situ studies we propose the mesangial cell as the culprit of the abnormal collagen production.
We excluded the involvement of the canine Col3A1 gene in the disease using simple segregation analysis. Mutations in the regulatory parts of the gene, or in the coding region, could have resulted in either increased production/expression of the gene or alternatively a changed structure that decreased the function or degradation of the protein with a secondary accumulation in the glomeruli. The studied haplotype included the regulatory and most of the coding parts of the gene. As the markers were localized exactly at the Col3A1-locus, the genetic distance from the markers to the functional mutation would be ∼0. The fact that all combinations of the parental haplotypes exist in diseased, as well as healthy offspring, proves that Col3A1 is not involved.
We also found increased presence of fibronectin occurring in a pattern similar to the distribution of the abnormal collagen, as has been previously seen in another dog with Col3GP.5 Fibronectin glomerulopathy in humans26 is another fibrillar glomerulopathy and a possible differential diagnosis. The organized deposits of fibronectin glomerulopathy consist of fibronectin that is PAS positive, electron-dense deposits by EM, in contrast to the weakly PAS-positive material containing cross-banded collagen fibrils in Col3GP. Accumulation of fibronectin in glomeruli was also demonstrated in some, but not all human cases of Col3GP.11, 18 Fibronectin is a multiadhesive glycoprotein of the extracellular matrix and has binding sites for collagens, with most avidity to collagen type III. The binding site of fibronectin to collagen corresponds to the binding site of mammalian collagenase to collagen, hence fibronectin may have a role in the regulation of collagen breakdown, and thus promote collagen accumulation.27 Furthermore, fibronectin binding to denaturized collagens is greater than to native molecules.27 In normal kidneys fibronectin is present in the mesangium, but a significant increase can be observed in most types of glomerulopathy, with a distribution related to the primary glomerular lesion.28 Hence it is likely that the fibronectin present in the glomeruli in our study is a secondary feature.
Immunohistochemistry also revealed that some of the mesangial cells in the glomeruli of affected cases were α-SMA positive, as previously seen in other dogs with Col3GP 5, 6 and in a human case.20 In another animal case report, however, α-SMA was not detected in affected glomeruli.3 Enhanced synthesis of α-SMA may be a marker of mesangial cell activation caused by injury,29, 30 and increased in conditions like nodular diabetic glomerulosclerosis and amyloidosis.29 It has been proposed that the activated mesangial cells may synthesize the collagen type III in human Col3GP.20 However, our Col3GP dogs showed only very few α-SMA-positive cells, making it unlikely that the extensive abnormal collagen deposition is only a result of mesangial cell activation.
Immunofluorescence studies showed irregular weak-positive staining for complement factor C3 and IgM in affected glomeruli, most likely resulting from non-specific glomerular trapping of these components. Glomerular C3 deposits were not detected in another described canine case where C3 was investigated, while glomerular IgM and IgG deposits were occasionally seen.6 In humans, immunofluorescence for IgG is mainly negative, and staining for C3 and IgM is negative or shows focal and segmental staining, often along the capillary walls,10, 15, 16, 17, 20, 31 coinciding with our findings. The deposits are described as non-specific2 and not indicative of an immune-mediated process,1 as also confirmed by lack of immune complexes by EM.
Another disease that is also characterized by glomerular accumulations of collagen type III, and a possible differential diagnosis, is the nail-patella syndrome (NPS). NPS is an autosomal dominant human disorder with glomerular, orthopedic and cutaneous manifestations, caused by mutations in the gene Lmx1b.32 By EM, collagen type III is present predominantly within the lamina densa of the GBM,2, 33 in contrast to the glomerulopathy occurring in this pedigree of dogs, with massive subendothelial and mesangial collagen accumulations.
Evidence of systemic disease was lacking in our Col3GP affected dogs, as neither extrarenal pathologic collagen nor other clinical or pathological signs of extrarenal disease related to Col3GP were observed. Human cases with collagen type III fibrils in one or more of the other internal organs of the patients have been reported,17, 34 and patients with Col3GP and concurrent anemia or respiratory symptoms have been described.11 Col3GP has been observed in two patients with factor H deficiency,2, 19 and a possible link between Col3GP and a defect in the complement system has been suggested. Our data support that Col3GP in this pedigree of dogs is a primary renal disease. However, only four of the dogs in our study developed clinical signs before euthanasia, and thus, more clinical studies are warranted. Except from confirming a variable deposition of complement in affected glomeruli, the complement systems of our dogs were not investigated in this study, neither were serum levels of procollagen III peptide, a non-invasive marker for the disease in humans.2 Transgenic mice with increased serum levels of interleukin-4 and transforming growth factor-β develop glomerulosclerosis with mesangial collagen accumulation,35, 36 and examining affected animals for elevated serum levels of these and other substances that might lead to collagen overproduction is of interest.
Our study demonstrates that the morphologic features of canine Col3GP are similar to Col3GP in humans, and the inheritance is autosomal recessive as also suggested in humans. Canine genetic models have previously been critical to the understanding of various genetic diseases in a number of tissues, including X-linked dominant hereditary nephritis (Alport syndrome), a genetically simple glomerular disease occurring spontaneously in dogs and humans,37 and most recently regarding the skin disorder congenital ichthyosis.38 Based on its similarities with the human condition, we propose that this canine autosomal recessive Col3GP may serve as a model to investigate the pathogenesis and the genetics of canine and human Col3GP.
In summary, we have herein described the morphologic features of canine autosomal Col3GP, the first animal model of Col3GP, and established the inheritance pattern by pedigree analysis. Dogs with this naturally acquired disease may facilitate the understanding of the nature of Col3GP in man and contribute to the development of a future treatment of this disease, and also illuminate fundamental mechanisms of excess collagen accumulation in other more common progressive diseases.
References
Alchi B, Nishi S, Narita I et al. Collagenofibrotic glomerulopathy: clinicopathologic overview of a rare glomerular disease. Am J Kidney Dis 2007;49:499–506.
Gubler MC, Heidet L, Antignac C . Alport’s Syndrome, Thin Basement Membrane Nephropathy, Nail-Patella Syndrome, and Type III Collagen Glomerulopathy. In: Jennette JC, Olson JL, Schwartz MM, et al. (eds) Heptinstall’s Pathology of the Kidney. 6th edn. Philadelphia: Lippincott Williams & Wilkins, 2007, pp 487–515.
Adachi K, Mori T, Ito T et al. Collagenofibrotic glomerulonephropathy in a cynomolgus macaque (Macaca fascicularis). Vet Pathol 2005;42:669–674.
Fujisama-Imura K, Takasu N, Tsuchiya N et al. Spontaneous collagenofibrotic glomerulonephropathy in a young cynomolgus monkey. J Toxicol Pathol 2004;17:279–282.
Kamiie J, Yasuno K, Ogihara K et al. Collagenofibrotic glomerulonephropathy with fibronectin deposition in a dog. Vet Pathol 2009;46:688–692.
Kobayashi R, Yasuno K, Ogihara K et al. Pathological characterization of collagenofibrotic glomerulonephropathy in a young dog. J Vet Med Sci 2009;71:1137–1141.
Koeman JP, Biewenga WJ, Gruys E . Proteinuria associated with glomerulosclerosis and glomerular collagen formation in three Newfoundland dog littermates. Vet Pathol 1994;31:188–193.
Shirota K, Masaki T, Kitada H et al. Renal glomerular fibrosis in two pigs. Vet Pathol 1995;32:236–241.
Nakamura S, Shibata S, Shirota K et al. Renal glomerular fibrosis in a cat. Vet Pathol 1996;33:696–699.
Tamura H, Matsuda A, Kidoguchi N et al. A family with two sisters with collagenofibrotic glomerulonephropathy. Am J Kidney Dis 1996;27:588–595.
Gubler MC, Dommergues JP, Foulard M et al. Collagen type III glomerulopathy: a new type of hereditary nephropathy. Pediatr Nephrol 1993;7:354–360.
Lium B, Moe L . Hereditary multifocal renal cystadenocarcinomas and nodular dermatofibrosis in the German shepherd dog: macroscopic and histopathologic changes. Vet Pathol 1985;22:447–455.
Lingaas F, Comstock KE, Kirkness EF et al. A mutation in the canine BHD gene is associated with hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis in the German Shepherd dog. Hum Mol Genet 2003;12:3043–3053.
Barthel LK, Raymond PA . Subcellular localization of alpha-tubulin and opsin mRNA in the goldfish retina using digoxigenin-labeled cRNA probes detected by alkaline phosphatase and HRP histochemistry. J Neurosci Methods 1993;50:145–152.
Ikeda K, Yokoyama H, Tomosugi N et al. Primary glomerular fibrosis: a new nephropathy caused by diffuse intra-glomerular increase in atypical type III collagen fibers. Clin Nephrol 1990;33:155–159.
Hisakawa N, Yasuoka N, Nishiya K et al. Collagenofibrotic glomerulonephropathy associated with immune complex deposits. Am J Nephrol 1998;18:134–141.
Mizuiri S, Hasegawa A, Kikuchi A et al. A case of collagenofibrotic glomerulopathy associated with hepatic perisinusoidal fibrosis. Nephron 1993;63:183–187.
Imbasciati E, Gherardi G, Morozumi K et al. Collagen type III glomerulopathy: a new idiopathic glomerular disease. Am J Nephrol 1991;11:422–429.
Vogt BA, Wyatt RJ, Burke BA et al. Inherited factor H deficiency and collagen type III glomerulopathy. Pediatr Nephrol 1995;9:11–15.
Naruse K, Ito H, Moriki T et al. Mesangial cell activation in the collagenofibrotic glomerulonephropathy. Case report and review of the literature. Virchows Arch 1998;433:183–188.
Ferreira RD, Custodio FB, Guimaraes CS et al. Collagenofibrotic glomerulopathy: three case reports in Brazil. Diagn Pathol 2009;4:33.
Williams BR, Gelman RA, Poppke DC et al. Collagen fibril formation. Optimal in vitro conditions and preliminary kinetic results. J Biol Chem 1978;253:6578–6585.
Scheinman JI, Brown DM, Michael AF . Collagen synthesis by human glomerular cells in culture. Biochim Biophys Acta 1978;542:128–136.
Foidart JM, Foidart JB, Mahieu PR . Synthesis of collagen and fibronectin by glomerular cells in culture. Ren Physiol 1980;3:183–192.
Razzaque MS, Koji T, Taguchi T et al. In situ localization of type III and type IV collagen-expressing cells in human diabetic nephropathy. J Pathol 1994;174:131–138.
Strom EH, Banfi G, Krapf R et al. Glomerulopathy associated with predominant fibronectin deposits: a newly recognized hereditary disease. Kidney Int 1995;48:163–170.
Minor RR . Collagen metabolism: a comparison of diseases of collagen and diseases affecting collagen. Am J Pathol 1980;98:225–280.
Dixon AJ, Burns J, Dunnill MS et al. Distribution of fibronectin in normal and diseased human kidneys. J Clin Pathol 1980;33:1021–1028.
Alpers CE, Hudkins KL, Gown AM et al. Enhanced expression of ‘muscle-specific’ actin in glomerulonephritis. Kidney Int 1992;41:1134–1142.
Johnson RJ, Iida H, Alpers CE et al. Expression of smooth muscle cell phenotype by rat mesangial cells in immune complex nephritis. Alpha-smooth muscle actin is a marker of mesangial cell proliferation. J Clin Invest 1991;87:847–858.
Ozu H, Nitta K, Yumura W et al. [A case of primary glomerular fibrosis associated with the accumulation of type I and type III collagen]. Nihon Jinzo Gakkai Shi 1994;36:1315–1319.
Dreyer SD, Zhou G, Baldini A et al. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet 1998;19:47–50.
Cohen AH . Collagen type III glomerulopathies. Adv Chronic Kidney Dis 2012;19:101–106.
Yasuda T, Imai H, Nakamoto Y et al. Collagenofibrotic glomerulopathy: a systemic disease. Am J Kidney Dis 1999;33:123–127.
Ruger BM, Erb KJ, He Y et al. Interleukin-4 transgenic mice develop glomerulosclerosis independent of immunoglobulin deposition. Eur J Immunol 2000;30:2698–2703.
Kopp JB, Factor VM, Mozes M et al. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest 1996;74:991–1003.
Lees GE, Helman RG, Kashtan CE et al. New form of X-linked dominant hereditary nephritis in dogs. Am J Vet Res 1999;60:373–383.
Grall A, Guaguere E, Planchais S et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat Genet 2012;44:140–147.
Acknowledgements
We wish to thank Lise Høgeli, Lars Ivar Løkken, Lene Hermansen and Inger Rudshaug for excellent technical assistance, and the Astri and Birger Torstedt Fund for the Benefit of Animals, for its economic support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The first animal model of collagen type III glomerulopathy with massive glomerular collagen type III deposition is described. Afflicted animals exhibit mesangial collagen type III synthesis, and genetic studies exclude mutations in the canine Col3A1 gene. This model may be useful for study of mechanisms of excess collagen accumulation.
Rights and permissions
About this article
Cite this article
Rørtveit, R., Lingaas, F., Bønsdorff, T. et al. A canine autosomal recessive model of collagen type III glomerulopathy. Lab Invest 92, 1483–1491 (2012). https://doi.org/10.1038/labinvest.2012.112
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2012.112
Keywords
This article is cited by
-
A clinical study of canine collagen type III glomerulopathy
BMC Veterinary Research (2013)
