
Abstract
The advent of phylogenetic DNA microarrays and high-throughput pyrosequencing technologies has dramatically increased the resolution and accuracy of detection of distinct microbial lineages in mixed microbial assemblages. Despite an expanding array of approaches for detecting microbes in a given sample, rapid and robust means of assessing the differential viability of these cells, as a function of phylogenetic lineage, remain elusive. In this study, pre-PCR propidium monoazide (PMA) treatment was coupled with downstream pyrosequencing and PhyloChip DNA microarray analyses to better understand the frequency, diversity and distribution of viable bacteria in spacecraft assembly cleanrooms. Sample fractions not treated with PMA, which were indicative of the presence of both live and dead cells, yielded a great abundance of highly diverse bacterial pyrosequences. In contrast, only 1% to 10% of all of the pyrosequencing reads, arising from a few robust bacterial lineages, originated from sample fractions that had been pre-treated with PMA. The results of PhyloChip analyses of PMA-treated and -untreated sample fractions were in agreement with those of pyrosequencing. The viable bacterial population detected in cleanrooms devoid of spacecraft hardware was far more diverse than that observed in cleanrooms that housed mission-critical spacecraft hardware. The latter was dominated by hardy, robust organisms previously reported to survive in oligotrophic cleanroom environments. Presented here are the findings of the first ever comprehensive effort to assess the viability of cells in low-biomass environmental samples, and correlate differential viability with phylogenetic affiliation.
Similar content being viewed by others

Introduction
Microbial cells are traditionally classified as either viable (maintaining active metabolism and membrane integrity), viable but dormant (because of external pressures) or non-viable (dead) (Kaprelyants et al., 1993; Keer and Birch, 2003). The vast majority of microorganisms cannot be cultivated and many that can require long cultivation times (Amann et al., 1995). To minimize the time spent in determining viability and bias associated with such analyses, advanced molecular approaches for assessing cellular viability have been recently developed, including live–dead staining (Boulos et al., 1999) and flow cytometry-based techniques (Ben-Amor et al., 2005). In such cases, however, there is inherent risk of overestimating the total number of viable cells in samples because of variation in binding affinities of the dyes used. Methods probing RNA as an alternative to DNA have also been developed for assessing viability (DeAngelis et al., 2011), but these too come with complications as RNA is difficult to purify, less stable, prone to degradation and often isolated in quantities insufficient for analysis (Hierro et al., 2006; Andorra et al., 2010).
The development of high-throughput pyrosequencing and phylogenetic microarray techniques has dramatically increased the resolution and detectable spectrum of diverse microbial lineages from environmental samples (Sogin et al., 2006; La Duc et al., 2009; Mendes et al., 2011). However, DNA-based molecular technologies alone have yet to be validated for assessing the differential viability of cells across varying phylogenetic lineages in a complex microbial assemblage. DNA-derived signals, which originate from both living and dead cell types, often lead to interpretations that overestimate the viable microbial population present (Rogers et al., 2008; Pointing et al., 2009). Quantitative PCR (qPCR)-based analyses increase the speed, sensitivity and specificity of quantitative microbial detection (Hierro et al., 2006; Yanez et al., 2011). Unfortunately, qPCR techniques alone cannot differentiate live from dead cells, as the latter contribute template DNA to the overall PCR amplification (Cawthorn and Witthuhn, 2008). The persistence of nucleic acids post-cell death renders cogent estimations of live–dead cellular ratios in a given sample practically impossible, as naked DNA will ultimately result in the overestimation of total cells (Nocker et al., 2006).
The treatment of microbial cell suspensions with propidium monoazide (PMA), which first intercalates and on photo-activation, covalently binds DNA, before DNA extraction followed by downstream PCR and related molecular analyses has become an increasingly popular technique for the selective detection and enumeration of viable microbes (Hein et al., 2006; Nocker et al., 2006; Wagner et al., 2008; Bae and Wuertz, 2009). The use of PMA for discriminating live from dead cells has been studied across various applications and research testbeds, including food (Cawthorn and Witthuhn, 2008), biosolids (van Frankenhuyzen et al., 2011), infectious enteric viruses in water samples (Parshionikar et al., 2010), fungi (Vesper et al., 2008), bacteriophage T4 (Fittipaldi et al., 2010), infectious parasitic protozoa (Brescia et al., 2009) and wastewater treatment plants (Lin et al., 2011). Recently, PMA treatment has been used in combination with crude microarray analysis for assessing cell viability (Nocker et al., 2009). In addition, pyrosequencing profiles of water samples exposed to high temperature and treated with and without PMA have been comparatively analyzed (Nocker et al., 2010).
The development and validation of molecular methods to selectively detect and enumerate the living fraction of the microbial population resident on critical surfaces (for example, hospital operating rooms, pharmaceutical manufacturing and packaging facilities, semiconductor fabrication, and spacecraft assembly cleanrooms) is of immense importance to good manufacturing practices aimed at minimizing contaminant bioburden levels. Reported here for the first time are the results of pioneering efforts coupling PMA-based viability discrimination with innovative PhyloChip DNA microarray and bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) methodologies to assess the viable bacterial population present in a typical low-biomass environmental sample.
Materials and methods
Sampling location
The two cleanroom facilities examined in this study were both certified as ISO 8. Spacecraft hardware and componentry was housed and assembled in the first cleanroom site, Jet Propulsion Laboratory’s (JPL’s) Spacecraft Assembly Facility (SAF) (sample #GI-36; Table 1), whereas the second cleanroom site, JPL cleanroom Building (Bldg)144 (sample #GI-42; Table 1), did not house spacecraft hardware at the time of sample collection. Both of these cleanroom facilities operated at a positive pressure, with temperatures in the range of 20±4 °C, and relative humidity ranging from 30% to 50%. Ground support equipment (GSE) consisted of all non-flight hardware items used during spacecraft hardware receipt, assembly, integration, test, storage, shipment and pre-launch activities. All GSE materials used inside the cleanrooms were inspected for compliance to visible cleanliness.
An all-purpose cleaning and degreasing agent (Kleenol 30, Accurate Industrial Supply, Inc., Cerritos, CA, USA, Cat #: J-CC-00040) was used to maintain cleanliness of the floor. Surface cleaning procedures were performed twice a day in the cleanroom during periods when spacecraft componentry was actively undergoing assembly (SAF; sample #GI-36). In contrast, the quiescent Bldg 144 environmental test facility certified cleanroom was cleaned only once a week since spacecraft hardware was not present in this facility at the time of sample collection. Both of the cleanroom facilities examined were maintained with stringent protocols pertaining to the replacement of tacky mats, vacuuming and mopping of floors, and wiping down of GSE surfaces with alcohol. In addition, before entering, staff were required to don cleanroom garments and comply with appropriate practices to minimize the influx of particulate matter.
Sample collection
Samples were collected from the cleanroom floors and GSE housed in SAF and Bldg 144. Wet-surface sampling of the cleanroom floors and GSE (each 1 m2) was performed using biological sampling kit (BiSKit; QuickSilver Analytics, Abingdon, MD, USA) as previously described (Kwan et al., 2011). To measure indigenous DNA associated with the sampling device (negative control), 15 ml of sterile phosphate-buffered saline (PBS, pH 7.0, Mo Bio Laboratories Inc., Carlsbad, CA, USA) was first processed through each BiSKit sampling module, with the expelled liquid pooled before using the very same kit for sample collection. The collection bottle corresponding to each BiSKit sampling module was replenished with 15 ml of sterile PBS and the entire assembly was inverted, saturating the macrofoam sponge. The sampling module of the BiSKit assembly was then used to sample floors and GSE sites in the ternary, unidirectional manner described elsewhere (Kwan et al., 2011). Overall, 36 individual samples (each 1 m2) were collected from two distinct cleanroom floors and two distinct GSE locations. A detailed description of the sample types is provided in Table 1. Sterile water, PBS, DNA extraction reagent blanks and PCR reagent blanks used in sample collection, processing and analysis, respectively, served as negative controls in all molecular analyses.
Sample processing
Sample volumes were extracted from BiSKit devices as instructed by the manufacturer, a total of three times with 15 ml of PBS each. Previous studies have demonstrated that spacecraft-associated surfaces house extremely low-biomass and seldom yield detectable PCR amplification products (Moissl et al., 2007; Vaishampayan et al., 2010). Hence, all samples collected from the same general location or site were pooled together. The biological materials resulting from each pooled sample (∼400 ml each) were further concentrated using Amicon Ultra-50 Ultracel centrifugal filter tubes (Millipore, Billerica, MA, USA). Each filter unit has a molecular mass cutoff of 50 kDa, which facilitates the concentration of bacterial cells, spores and exogenous nucleic acid fragments >100 bp in a final volume of 1 ml. All filtered samples were divided into two separate aliquots (500 μl each), one to be subjected to PMA pre-treatment (viability assessment), and the other to serve as a null environmental sample (viable+non-viable; total DNA). All samples, both with and without PMA pre-treatment were subjected to DNA extraction via the Maxwell 16 automated system (Promega, Madison, WI, USA) according to the manufacturer’s instructions, and resulting DNA suspensions (100 μl each) were stored at −20 °C.
PMA treatment
A 500 μl aliquot of filter-concentrated sample suspension was treated with 12.5 μl of PMA (2 mM; Biotium, Inc., Hayward, CA, USA) to a final concentration of 50 μM (Rawsthorne et al., 2009; Nocker et al., 2010), followed by thorough mixing and incubation in the dark for 50 min at room temperature. The tubes were inverted manually 5–6 times over a 10-min incubation interval to promote homogeneous PMA exposure. Sample tubes were then placed horizontally atop a bed of ice and exposed to a 500-W halogen lamp (Osram 64553 C318; Danvers, MA, USA) at a distance of 20 cm for 3 min. Samples without PMA treatment were also subjected to incubation in dark for 50 min and exposure to 500-W halogen lamp on ice along with PMA-treated samples.
Quantitative PCR
Bacteria-directed primers (1369F and 1492R) targeting the 16S ribosomal RNA (rRNA) gene were used for qPCR analysis (Suzuki et al., 2000). Each 25 μl reaction mixture consisted of 12.5 μl of Bio-Rad 2X iQ SYBR Green Supermix (Hercules, CA, USA), 10.5 μl of UltraPure water (Gibco; Grand Island, NY, USA), 0.5 μl of forward primer 1369F (10 μM), 0.5 μl of reverse primer 1492R (10 μM) and 1 μl of DNA template. Purified standards and UltraPure Gibco water no-template controls were included in all qPCR runs. Thermal cycling parameters for universal 16S rRNA gene qPCR were as follows: hold at 95 °C for 3 min to achieve initial denaturation, followed by 40 cycles of: 10-s hold at 95 °C to denature, ramp-down to 55 °C for primer annealing and extension occurring through a 35-s ramp-up to 95 °C. In this study, all samples were analyzed in triplicate.
Taxonomy
For convenience and differentiation in this communication, bTEFAP-based pyrosequence discrimination results are binned hierarchically into what are referred to as molecular operational taxonomic unit(s) (MOTU; Blaxter, 2003; Blaxter et al., 2005). Variation in DNA sequence among microbes can arise via naturally occurring evolutionary events and/or methodological errors (for example, homopolymer repetition in pyrosequencing). It is the goal of the MOTU-based classification and clustering system presented here, and in detail elsewhere (Blaxter and Floyd, 2003), to separate these two sources of sequence variation based on known error rates in sequencing and measured levels of difference across various taxonomical schemes. The accuracy and specificity of a MOTU-based system can be derived from measured levels of between-taxa and within-group variation from well-defined populations, and of observational error obtained by re-sequencing (Blaxter and Floyd, 2003). In a similar vein, PhyloChip DNA microarrays use multiple ∼25-bp probes, which collectively represent the full-length ∼1.5-kb 16S rRNA gene of each taxon. In this study, PhyloChip-derived taxonomic units (PTUs) were delineated in accordance with the hybridization scores of a given set of 25-mer probes, which have been previously designed based on the prevalence of members of a given PTU, and dissimilarity in DNA sequences outside of the given PTU. Ultimately, a microorganism can be assigned to only one given MOTU/PTU, either via similarity within a homologous sequenced DNA fragment (MOTU) or hybridization score (PTU), but neither MOTU nor PTU need be congruent with other taxonomic schemes.
PhyloChip G3
Bacterial 16S rRNA genes were amplified from DNA preparations from each sample using the primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′). PCR conditions were as follows: 1 cycle of initial melting for 3 min at 95 °C, followed by 35 cycles of 30-s melting at 95 °C, 30-s annealing over a 48–58 °C gradient (48 °C, 48.8 °C, 50.1 °C, 51.9 °C, 54.4 °C, 56.3 °C, 57.5 °C and 58 °C), and 2-min extension at 72 °C, with a final 10-min incubation at 72 °C. The amount of total 16S rRNA gene PCR product subjected to hybridization on PhyloChips was normalized across samples (∼400 ng) whenever possible. A detailed explanation of the processing of the PhyloChip assay has been described elsewhere (Hazen et al., 2010). Stage 1 and stage 2 analysis were performed and the cross-hybridization response score was adjusted as previous described (Cooper et al., 2011).
PhyloChip G3 data analysis
Following stage 2 analysis, hybridization intensities were transformed (log2*1000) and were henceforth referred to as transformed hybridization intensities. Representative PTU sequences were then compared against the taxonomic architecture of the SILVA database (Pruesse et al., 2007) and PTU were grouped at the genus level. In order to be identified as having been enriched in the PMA-treated samples, PTU had to meet the following two criteria: (a) the PTU must be deemed present based on standard PhyloChip analysis in PMA-treated samples and (b) the corresponding PTU must be present in greater transformed hybridization intensity in the PMA-treated sample fraction than in the non-PMA-treated sample fraction.
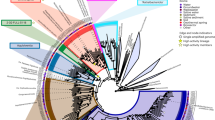
One representative 16S rRNA gene sequence within each PTU was selected, a multiple sequence alignment was generated with the SINA aligner (Pruesse et al., 2012), and a neighbor-joining phylogenetic tree was compiled at the family level with MEGA 4 (Tamura et al., 2007). This tree was rendered in a circular manner with the iTOL tree viewing program (Letunic and Bork, 2011).
Tag-encoded FLX amplicon pyrosequencing
Bacterial-biased primers 28F (5′-GAGTTTGATCNTGGCTCAG-3′) and 519R (5′-GTNTTACNGCGGCKGCTG-3′) were used to amplify ∼500-bp fragments spanning the V1–V3 hypervariable regions of the bacterial 16S rRNA gene. This primer pair was tailored for bTEFAP by adding a fusion linker and a proprietary 12-bp barcode sequence at the 5′ end of the forward primer, and a biotin and fusion linker sequence at the 5′ end of the reverse primer (Dowd et al., 2008). A HotStarTaq Plus master mix kit (Qiagen, Valencia, CA, USA) was used to catalyze the PCR under the following thermal cycling conditions: initial denaturing at 95 °C for 5 min, followed by 35 cycles of denaturing at 95 °C for 30 s, annealing at 54 °C for 40 s, and extension at 72 °C for 1 min, finalized by a 10-min elongation at 72 °C. Resulting PCR products were purified via Rapid Tip (Diffinity Genomics, Inc., West Henrietta, NY, USA) chemistry, and were then pooled accordingly. Small fragments (<100 bp) were removed with Agencourt Ampure Beads in accordance with manufacturer’s instructions (Beckman Coulter, Brea, CA, USA).
In preparation for FLX-Titanium sequencing (Roche, Nutley, NJ, USA), resulting PCR amplicon fragment size and concentration were accurately measured with DNA 1000 chips using a Bioanalyzer 2100 automated electrophoresis station (Agilent, Santa Clara, CA, USA) and a TBS-380 Fluorometer (Turner Biosystems, Sunnyvale, CA, USA). The total volume of initial PCR product used for subsequent emulsion PCR was 2 μl for strong positives (>10 ng μl−1), 5 μl for weak positives (5 to 10 ng μl−1) and 20 μl for samples that failed to yield PCR products (<5 ng μl−1). This normalization step helped to ensure minimal bias favoring downstream amplification from initially strong PCR products. Approximately 9.6 × 106 molecules of ∼600-bp double-stranded DNA were combined with 9.6 × 106 DNA capture beads, and then subjected to emulsion PCR conditions. Following recovery and enrichment, bead-attached DNA molecules were denatured with NaOH and sequencing primers were annealed. A 454 pyrosequencing run was performed on a GS PicoTiterPlate using the Genome Sequencer FLX System in accordance with manufacturer’s instructions (Roche). In all, 24 to 30 tagged samples were applied to each quarter region of the PicoTiterPlate. All bTEFAP procedures were performed at the Research and Testing Laboratory (Lubbock, TX, USA) in accordance with well-established protocols (Dowd et al., 2008).
bTEFAP-derived bacterial diversity and data analysis
Bacterial TEFAP sequences were processed and analyzed using the MOTHUR software package (Schloss et al., 2009), with the AmpliconNoise algorithm implemented. Raw pyrosequencing data for the PMA-untreated samples was derived from previously published work and was re-analyzed alongside PMA-treated samples (La Duc et al., 2012). Previously described standard operating procedures were followed for the analysis of sequence data in this study (Schloss et al., 2011). Sequences were removed from consideration if they (a) did not contain the primer sequence, (b) contained an uncorrectable barcode, (c) were <200 nt in length, (d) had homopolymers longer than 8 nt or (e) had a quality score of <25. Unique sequences were aligned using the Greengenes reference alignment (Schloss et al., 2009; McDonald et al., 2012) and trimmed such that all the sequences overlap in the same alignment space. Filtered sequences were assigned to samples according to their 12-nt barcode. After removing chimeras, sequences were classified in accordance with the new Greengenes training set and taxonomy (McDonald et al., 2012; Werner et al., 2012), and clustered into MOTU at the 0.03 level (that is, at 97% similarity) (Schloss et al., 2011).
Negative controls
During this study, a negative control whereby a BiSKit was only pre-moistened with PBS, and a handling control, in which a BiSKit was pre-moistened with PBS and exposed to the sampling environment, were also prepared. These negative and handling controls were processed and analyzed both with and without PMA treatment before DNA extraction. All of the resulting qPCR indices for these controls were below detection limit. Bacterial bTEFAP sequencing was not performed on any of these controls because PCR amplification did not yield any quantifiable product. Although no detectable PCR amplification products were available, all negative and handling controls were run on a PhyloChip in order to detect possible contaminants. The resulting 447 PTU (of 8943 PTU in total) detected were omitted from the entire analysis. Only one PTU was detected in one handling control after PMA treatment. No PTU were detected in any of the negative or sampling controls after PMA treatment, supporting the conclusion that the detected PTU originated from extraneous DNA, and not from viable microbes associated with sampling materials or reagents.
Statistical analysis of community data
Multiple statistical analyses were performed to study the differences between the PMA-treated and non-PMA samples, all of which were based on (a) the abundance of sequences of each MOTU and (b) the transformed hybridization intensities of each PTU. This included principal coordinate analysis (PCoA), multi-response permutation procedures and Adonis testing (999 permutations), all of which were based on Bray–Curtis distance measures. Dendogram clustering was based on Euclidean distance. Diversity indices (Shannon–Wiener) were calculated for MOTU only. All statistical analyses, including heatmaps, were performed using the R programming environment (R-project 2011, Vegan, MASS and ape packages).
Results
Quantitative PCR
Total bacterial burden, as assessed by bacteria-directed qPCR, is given in Table 1. When PMA treatment was omitted before molecular processing, the total bacterial burden (viable+non-viable) of the mission-critical SAF cleanroom floor and GSE samples was ∼105 to 106 rRNA gene copies m−2. Samples collected from the inactive Bldg 144 facility floor and GSE yielded 1 to 2-logs higher rRNA gene copy numbers than SAF samples both with and without PMA treatment. Following treatment with PMA, a mere 7% (floors) and 4% (GSE) of the total bacterial population encountered about the mission-critical SAF was determined to be viable. However, the bacterial population present in PMA-treated Bldg 144 samples was 21% (floors) and 7% (GSE) viable. On comparative analysis, Bldg 144 floors housed a 188-fold greater viable bacterial burden than their mission-critical SAF counterparts. Similarly, Bldg 144 GSE samples contained 25-fold more viable bacteria than their SAF GSE equivalents.
bTEFAP analysis
A breakdown of the number of MOTU observed in the various samples examined over the course of this study is provided in Table 1. Overall, Bldg 144 cleanroom floor samples housed 3.6-fold more bacterial MOTU than the mission-critical SAF cleanroom floor, whereas the GSE surfaces from each of these cleanrooms yielded a roughly equivalent number of MOTU. The effect of PMA treatment was significantly higher in both SAF (97% MOTU reduction) and Bldg 144 (75% MOTU reduction) floor samples, which indicated the presence of a large number of dead cells or extraneous DNA on these floors. A similar trend was observed in PMA-treated GSE samples from SAF (reduced from 425 to 17 MOTU) and Bldg 144 (reduced from 571 to 42 MOTU).
A breakdown of MOTU, at the level of bacterial phyla or class, observed in the various samples examined is detailed in Table 2. The mission-critical SAF floor retained MOTU affiliated with physiologically recalcitrant bacteria (Actinobacteria, Acidobacteria and Firmicutes), whereas the Bldg 144 cleanroom floor harbored predominantly proteobacterial MOTU. It was particularly apparent that a few acidobacterial types (4 MOTU) were present in great abundance (112 sequences) in the SAF floor samples. However, the GSE samples exhibited no such correlation between MOTU numbers and sequence occurrence. A closer examination of the pyrosequence reads resulting from the SAF cleanroom floor and GSE samples indicated a predominance of members of the genera: Acidobacteria, Actinobacteria, Arsenicicoccus, Arthrobacter, Corynebacterium, Kineococcus, Propionibacterium, Nocardioides, Streptomyces, Bacillus, Clostridium, Lactobacillus, Deinococcus and Staphylococcus. Many of the sequences arising from the physiologically recalcitrant bacteria observed in cleanroom floor samples sans PMA treatment were absent or in very low number in the PMA-treated fractions of the very same sample.
Although equivalent surface areas were sampled from the floors and GSE of the two cleanrooms studied (9 m2 each), the Bldg 144 samples gave rise to many more pyrosequence reads than the SAF floor samples. Regardless of sample type, PMA-treated sample fractions consistently yielded considerably fewer pyrosequences than their untreated counterparts. Anywhere from 14 to 2196 high-quality pyrosequences (>250 bp) were obtained from samples that had been pre-treated with PMA, whereas 1783 to 15 914 high-quality pyrosequences were recovered from untreated samples. Even when PMA treatment was omitted, the mission-critical SAF cleanroom floor sample (GI-36-4) yielded far fewer pyrosequences (1783 reads) than the Bldg 144 cleanroom floor sample (GI-42-1; 15 914 reads). The relative abundance of pyrosequences retrieved from the PMA-untreated cleanroom floor samples is plotted as a Venn diagram in Figure 1a. Approximately 65% of the pyrosequences retrieved from these two distinct facility floor samples were detected in both cleanrooms, although this shared fraction represented only 8% of the total observed MOTU (46 out of 569; Figure 1a). The relative abundance of pyrosequences retrieved from the untreated SAF samples is plotted as a Venn diagram in Figure 1b. Between the SAF floor and GSE samples, ∼38% of the total number of detected pyrosequence reads were shared, although this constituted a mere 7.8% of the total MOTU (43 out of 547).

Venn diagram showing the MOTU detected in various samples of the cleanrooms. Comparison (a) between SAF and Bldg 144 cleanroom floor samples and (b) between floor and GSE samples of SAF cleanroom. Parentheses denote total number of pyrosequences generated and the numerals without parentheses are total number of MOTU present in that sample.
PhyloChip analysis
A drastic decrease in the total number of bacterial genera was observed in all samples on pre-treatment with PMA (Table 1). After having been treated with PMA, the SAF cleanroom floor and GSE samples exhibited very simple bacterial community structure, housing very few genera (9 and 3, respectively) of Firmicute and Proteobacteria lineage. Without PMA treatment, these very same floor and GSE samples yielded many more genera, which represented a diverse assemblage of taxa (Table 3) dominated by Actinobacteria, Firmicutes and Proteobacteria. The Bldg 144 cleanroom floor and GSE samples also presented many more genera without PMA pre-treatment (199 and 236, respectively) than the very same samples having been treated with PMA (106 and 75).
A heatmap was generated based on the transformed hybridization intensities of detected PTU (Supplementary Figure 1). Such analysis clearly illustrates a marked decrease in the hybridization scores of all samples treated with PMA, as compared with the very same samples not treated with PMA. For instance, the SAF GSE sample (GI-36-3) exhibited very high transformed hybridization intensities for detected PTU sans PMA treatment. However, when treated with PMA, this very same sample showed a dramatic reduction in all resulting hybridization intensity scores, which suggested that the majority of the detected 16S rRNA genes arose from deceased members of the community. In order to identify the viable members that exhibited increased hybridization intensities after PMA treatment, ratios of non-PMA-treated and PMA-treated hybridization scores were calculated for each PTU, and are presented as a heatmap (Figure 2). An observed increase in the transformed hybridization intensity of a given PTU following PMA treatment would likely stem from PCR bias in non-PMA-treated samples, where high levels of DNA originating from non-viable cells are co-amplified. This amplification of template DNA arising from non-viable cell types masks the presence of smaller levels of DNA template arising from viable cells. Hence, transformed hybridization intensities are elevated on removing the template DNA arising from non-viable cells from the equation (that is, PMA-treated sample fraction). The SAF cleanroom floor and GSE samples gave rise to fewer PTU with higher transformed hybridization ratios. At the same time, PMA-treated Bldg 144 cleanroom samples were richer in PTU having increased transformed hybridization intensity ratios, which was indicative of a relatively greater viable population. These PTU belonged to the Firmicutes, Cyanobacteria, Actinobacteria and Proteobacteria. A phylogenetic tree of PhyloChip PTU grouped at the family level is provided as Supplementary Figure 2, which shows the presence of the detected families in each sample with and without PMA treatment.

Heatmap of PTU that increased in transformed hybridization intensities in PMA-treated samples compared with non-PMA-treated samples and were called present in the PMA-treated sample. An increase in transformed hybridization intensities in PMA-treated sample is reflected as a positive ratio. In total, 801 PTU were identified that fulfilled this requirement, which were grouped into 70 genera. Displayed are representatives of all genera with the most drastic changes for each sample pair. Numbers in parentheses are the total number of PTU.
Statistical analysis of microbial community profiles
The various samples examined by bTEFAP analysis consistently showed lower Shannon–Wiener diversity indices when treated with PMA compared with their corresponding non-treated samples (Supplementary Table 1). PCoA was performed to study the environmental clustering and relatedness of community profiles derived from bTEFAP and PhyloChip analyses (Figure 3). All of the samples analyzed without PMA treatment clustered together, indicative of their relatively similar community structure, compared with their coinciding PMA-treated sample fractions. The clustering observed in the PCoA plot was congruent with the dendogram clustering presented in Supplementary Figure 1, which implied a close association among all non-PMA samples. Multi-response permutation procedure analysis was performed to assess the difference in community structure between PMA-treated sample fractions and non-PMA-treated sample fractions, derived by both bTEFAP and PhyloChip analysis. The null hypothesis (no difference in PMA-treated and non-PMA-treated samples) was rejected based on the significance of the delta for both bTEFAP and PhyloChip analyses (0.028 and 0.037, respectively). The chance-corrected within-group agreements were fairly low for bTEFAP sequence data (A=0.0411) but high (A=0.2605) for PhyloChip data, which reflect the observed grouping in PCoA. In addition, Adonis testing clearly showed a significant change in the detected community profiles after PMA treatment (P-value bTEFAP: 0.04, P-value PhyloChip: 0.02). Based on the multiple statistical approaches used, such as Adonis, multi-response permutation procedure, a Euclidean distance-based dendogram (Supplementary Figure 1), and ordination (PCoA) analyses (Figure 3), it was clear that PMA-treated samples and non-PMA samples were significantly dissimilar with respect to diversity in bacterial community profiles.

PCoA based on: (a) number of pyrosequences per MOTU (PCoA1, percentage of explained variance: 22%; PCoA2, percentage of explained variance: 17%) and (b) PhyloChip-derived transformed hybridization scores of each PTU (PCoA1, percentage of explained variance: 87%; PCoA2, percentage of explained variance: 6%). Open and closed dots represent PMA-treated and PMA-non-treated samples, respectively.
Discussion
Over the past 25 years, sequence analysis of PCR-amplified rRNA genes has become the ‘gold standard’ for assessing species richness in mixed microbial communities, and as a result, total resolvable microbial diversity is now estimated to be threefold greater than that based solely on cultivation (Pace, 1997). As sequences from organisms in greatest abundance are far more likely to be represented in clone libraries than those from singleton and low-abundance taxa, it is advantageous to use high-throughput technologies, such as bTEFAP and PhyloChip, which have been shown to render a far superior representation of community structure (Brodie et al., 2006; Sogin et al., 2006; DeSantis et al., 2007; La Duc et al., 2009).
The application of PMA to assess the differential viability of microbial cells is increasing in popularity within the scientific community (Nocker et al., 2009; van Frankenhuyzen et al., 2011). When used to pre-treat samples, the PMA concentration applied in this study effectively precluded ∼90% of the total DNA template molecules from downstream manipulation, resulting in the generation of only 2827 pyrosequences, whereas 27 449 pyrosequences were obtained from the very same samples without PMA pre-treatment. With respect to diversity, all of the pyrosequences generated after PMA treatment represented a mere 171 MOTU, which corresponded to ∼12% of the total number of MOTU resulting from the very same samples without PMA treatment (Table 2). Furthermore, 2- to 3-logs fewer pyrosequences arose from PMA-treated than untreated mission-critical cleanroom samples (0.8% for floor; 2.1% for GSE).
The total number of MOTU observed in the operational mission-critical SAF cleanroom floor sample (122 MOTU) was considerably less than that associated with the quiescent Bldg 144 cleanroom floor (447 MOTU). Bioinformatic analyses of pyrosequence data demonstrated that both of these contaminant microbial populations comprised only Actinobacteria, Firmicutes and Proteobacteria. The vast majority of these bacteria, if not all, were present in the SAF mission-critical cleanroom floor samples in a non-viable state. However, about 25% of the detected MOTU (108 out of 447) were observed to be viable in the Bldg 144 facility, suggesting that certain taxa are able to withstand the desiccated and nutrient-deprived conditions of these cleanroom floors (sample #GI-42-1). Actinobacterial genera such as Kineococcus, Kocuria, Modestobacter and Propionibacterium were present in high abundance in the JPL-SAF floor samples. In contrast, the floors of the Bldg 144 facility housed predominantly Proteobacterial genera, as Brevendimonas and Acinetobacter pyrosequences were generated in great numbers. Previously, members of these and other closely related genera have been isolated from spacecraft assembly environments (Osman et al., 2008; La Duc et al., 2009; Ghosh et al., 2010; Vaishampayan et al., 2012). The molecular biological detection and isolation of these robust microbial lineages from spacecraft-associated environments is of particular consequence to National Aeronautics and Space Administration (NASA) planetary protection practices, not to mention routine validation of these cleanroom facilities. Unlike cleanroom floors, which were treated with Kleenol 30 detergent, the GSE materials housed in either cleanroom were subjected only to alcohol wiping, and yet gave rise to similar MOTU and pyrosequence occurrence, with GSE materials kept at the Bldg 144 facility marginally enriched (∼1.3-fold increase in pyrosequences and MOTU). From these results, it is apparent that GSE need to be subjected to more rigorous cleaning regimens, as GSE-associated richness was two to four times greater than that of the floor surfaces.
The most frequently encountered bacterial MOTU from the stringently maintained and frequently cleaned floors of the SAF cleanroom were members of the genera: Bacillus, Clostridium and Nocardia. These bacteria are known to survive oligotrophic conditions for extended periods of time, tolerate alkaline and oxidative stress, and avoid death by ultraviolet radiation by morphing into highly resilient, dormant endospores (La Duc et al., 2007; Ghosh et al., 2010; Probst et al., 2010). Although routine cleaning and maintenance regimens limit the number of bacterial taxa capable of persisting in cleanrooms, hardy spore-forming microorganisms like Bacillus spp., Clostridium spp. and Nocardia spp. capitalize on their selective advantage and superior fitness and survive—much to the chagrin of those challenged with bioreduction and sterilization of these environments. The presence of these organisms is detected as a result of their (a) viability, or (b) inability to be penetrated by PMA molecules while in a non-viable state. Owing to subtle nuances inherent in the PMA-chemistry-coupled techniques described herein, endospores and non-viable cells having intact cell walls and/or outer membranes will escape PMA treatment (Nocker et al., 2009; Rawsthorne et al., 2009; Probst et al., 2012), and thus be observed as false-positive viable entities. Similarly, sampling and sample processing steps (for example, the composition of the solution to collect and store microorganisms, method for cell concentration and PMA treatment) might affect the viability of the cells but in a recent study such adverse effect of sample handling procedures was not noticed for sea and canal water samples (Kort et al., 2010; Nocker et al., 2010).
As was observed via bTEFAP procedures, PhyloChip-based analyses discerned noticeable differences in the bacterial diversity profiles resulting from PMA- and non-PMA-treated samples. One such observation was the reduction in diversity of proteobacteria detected in PMA-treated samples and prevalence of proteobacteria in samples not treated with PMA. Another consistent result was the observed prevalence of Firmicutes in the PMA-treated samples. This suggests that Gram-positive bacteria are more tolerable of the inhospitable conditions of the cleanroom environment that their Gram-negative kin.
Numerous studies have reported varying accounts of the microbial diversity typical of spacecraft-associated cleanrooms, and what the presence of such communities might portend for life detection endeavors in extraterrestrial settings (Venkateswaran et al., 2001; La Duc et al., 2004, 2007; Moissl et al., 2007; Probst et al., 2010; Vaishampayan et al., 2010). Indeed, the introduction of contaminant microbes to extraterrestrial environments could have profound repercussions on (a) the scientific integrity of in situ and sample-return based life detection experiments, and (b) the uncompromised nature of such settings. The worst-case scenario for life-detection experimentation would be the inadvertent transfer of viable contaminant microbiota to an otherwise pristine location of interest. The results of this study are encouraging, as they suggest that hitherto, the breadth of diversity enveloped within the viable fraction(s) of typical spacecraft-associated microbial communities have been overestimated. At the same time, these findings enabled the first ever statistically significant differentiation between the total and viable-only portion of microbial communities in cleanroom environments. Significant differences were shown between these two populations using two independent profiling methods, namely bTEFAP and PhyloChip G3. Consequently, these methodologies are an attractive means of discerning viable phylotypes in low-biomass environments. Such a capability is of crucial importance and benefit to numerous industries (for example, healthcare, pharma, semiconductor fabrication), not least of all the NASA, whose planetary protection program is tasked with ensuring spacecraft-borne microorganisms do not result in harmful contamination of extraterrestrial environments.
References
Amann RI, Ludwig W, Schleifer KH . (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59: 143–169.
Andorra I, Esteve-Zarzoso B, Guillamon JM, Mas A . (2010). Determination of viable wine yeast using DNA binding dyes and quantitative PCR. Int J Food Microbiol 144: 257–262.
Bae S, Wuertz S . (2009). Discrimination of viable and dead fecal Bacteroidales bacteria by quantitative PCR with propidium monoazide. Appl Environ Microbiol 75: 2940–2944.
Ben-Amor K, Heilig H, Smidt H, Vaughan EE, Abee T, de Vos WM . (2005). Genetic diversity of viable, injured, and dead fecal bacteria assessed by fluorescence-activated cell sorting and 16S rRNA gene analysis. Appl Environ Microbiol 71: 4679–4689.
Blaxter M . (2003). Molecular systematics: counting angels with DNA. Nature 421: 122–124.
Blaxter M, Floyd R . (2003). Molecular taxonomics for biodiversity surveys: already a reality. TRENDS Ecol Evol 18: 268–269.
Blaxter M, Mann J, Chapman T, Thomas F, Whitton C, Floyd R et al. (2005). Defining operational taxonomic units using DNA barcode data. Philos Trans R Soc Lond B Biol Sci 360: 1935–1943.
Boulos L, Prévost M, Barbeau B, Coallier J, Desjardins R . (1999). LIVE/DEAD® BacLight™: application of a new rapid staining method for direct enumeration of viable and total bacteria in drinking water. J Microbiol Methods 37: 77–86.
Brescia CC, Griffin SM, Ware MW, Varughese EA, Egorov AI, Villegas EN . (2009). Cryptosporidium propidium monoazide-PCR, a molecular biology-based technique for genotyping of viable Cryptosporidium oocysts. Appl Environ Microbiol 75: 6856–6863.
Brodie EL, DeSantis TZ, Joyner DC, Baek SM, Larsen JT, Andersen GL et al. (2006). Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl Environ Microbiol 72: 6288–6298.
Cawthorn DM, Witthuhn RC . (2008). Selective PCR detection of viable Enterobacter sakazakii cells utilizing propidium monoazide or ethidium bromide monoazide. J Appl Microbiol 105: 1178–1185.
Cooper M, La Duc MT, Probst A, Vaishampayan P, Stam C, Benardini JN et al. (2011). Comparison of innovative molecular approaches and standard spore assays for assessment of surface cleanliness. Appl Environ Microbiol 77: 5438–5444.
DeAngelis KM, Wu CH, Beller HR, Brodie EL, Chakraborty R, DeSantis TZ et al. (2011). PCR amplification-independent methods for detection of microbial communities by the high-density microarray PhyloChip. Appl Environ Microbiol 77: 6313–6322.
DeSantis TZ, Brodie EL, Moberg JP, Zubieta IX, Piceno YM, Andersen GL . (2007). High-density universal 16S rRNA microarray analysis reveals broader diversity than typical clone library when sampling the environment. Microb Ecol 53: 371–383.
Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA et al. (2008). Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 8: 43.
Fittipaldi M, Codony F, Adrados B, Camper AK, Morato J . (2010). Viable real-time PCR in environmental samples: can all data be interpreted directly? Microb Ecol 61: 7–12.
Ghosh S, Osman S, Vaishampayan P, Venkateswaran K . (2010). Recurrent isolation of extremotolerant bacteria from the clean room where Phoenix spacecraft components were assembled. Astrobiology 10: 325–335.
Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, Singh N et al. (2010). Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science 330: 204–208.
Hein I, Flekna G, Wagner M, Nocker A, Camper AK . (2006). Possible errors in the interpretation of ethidium bromide and PicoGreen DNA staining results from ethidium monoazide-treated DNA. Appl Environ Microbiol 72: 6860–6861 author reply 6861-6862.
Hierro N, Esteve-Zarzoso B, Gonzalez A, Mas A, Guillamon JM . (2006). Real-time quantitative PCR (QPCR) and reverse transcription-QPCR for detection and enumeration of total yeasts in wine. Appl Environ Microbiol 72: 7148–7155.
Kaprelyants AS, Gottschal JC, Kell DB . (1993). Dormancy in non-sporulating bacteria. FEMS Microbiol Rev 10: 271–285.
Keer JT, Birch L . (2003). Molecular methods for the assessment of bacterial viability. J Microbiol Methods 53: 175–183.
Kort R, Nocker A, de Kat Angelino-Bart A, van Veen S, Verheij H, Schuren F et al. (2010). Real-time detection of viable microorganisms by intracellular phototautomerism. BMC Biotechnol 10: 45.
Kwan K, Cooper M, Duc MTL, Vaishampayan P, Stam C, Benardini JN et al. (2011). Evaluation of procedures for the collection, processing, and analysis of biomolecules from low-biomass surfaces. Appl Environ Microbiol 77: 2943–2953.
La Duc MT, Kern RG, Venkateswaran K . (2004). Microbial monitoring of spacecraft and associated environments. Microb Ecol 47: 150–158.
La Duc MT, Dekas AE, Osman S, Moissl C, Newcombe D, Venkateswaran K . (2007). Isolation and characterization of bacteria capable of tolerating the extreme conditions of clean-room environments. Appl Environ Microbiol 73: 2600–2611.
La Duc MT, Osman S, Vaishampayan P, Piceno Y, Andersen G, Spry JA et al. (2009). Comprehensive census of bacteria in clean rooms by using DNA microarray and cloning methods. Appl Environ Microbiol 75: 6559–6567.
La Duc MT, Vaishampayan P, Nilsson H, Torok T, Venkateswaran K . (2012). Pyrosequencing-derived bacterial, archaeal, and fungal diversity of spacecraft hardware destined for Mars. Appl Environ Microbiol 78: 5912–5922.
Letunic I, Bork P . (2011). Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39: W475–W478.
Lin W-T, Luo J-F, Guo Y . (2011). Comparison and characterization of microbial communities in sulfide-rich wastewater with and without propidium monoazide Treatment. Curr Microbiol 62: 374–381.
McDonald D, Price MN, Goodrich J, Nawrocki EP, Desantis TZ, Probst A et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6: 610–618.
Mendes R, Kruijt M, de Bruijn I, Dekkers E, van der Voort M, Schneider JH et al. (2011). Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332: 1097–1100.
Moissl C, La Duc MT, Osman S, Dekas AE, Venkateswaran K . (2007). Molecular bacterial community analysis of clean rooms where spacecraft are assembled. FEMS Microbiol Ecol 61: 509–521.
Nocker A, Cheung CY, Camper AK . (2006). Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J Microbiol Methods 67: 310–320.
Nocker A, Mazza A, Masson L, Camper AK, Brousseau R . (2009). Selective detection of live bacteria combining propidium monoazide sample treatment with microarray technology. J Microbiol Methods 76: 253–261.
Nocker A, Richter-Heitmann T, Montijn R, Schuren F, Kort R . (2010). Discrimination between live and dead cellsin bacterial communities from environmental water samples analyzed by 454 pyrosequencing. Int Microbiol 13: 59–65.
Osman S, Peeters Z, LaDuc MT, Mancinelli R, Ehrenfreund P, Venkateswaran K . (2008). Effect of shadowing on the survival of bacteria to conditions simulating Martian atmosphere and UV-radiation. Appl Environ Microbiol 74: 959–970.
Pace NR . (1997). A molecular view of microbial diversity and the biosphere. Science 276: 734–740.
Parshionikar S, Laseke I, Fout GS . (2010). Use of propidium monoazide in reverse transcriptase PCR to distinguish between infectious and noninfectious enteric viruses in water samples. Appl Environ Microbiol 76: 4318–4326.
Pointing SB, Chan Y, Lacap DC, Lau MC, Jurgens JA, Farrell RL . (2009). Highly specialized microbial diversity in hyper-arid polar desert. Proc Nat Acad Sci Usa 106: 19964–19969.
Probst A, Vaishampayan P, Osman S, Moissl-Eichinger C, Andersen GL, Venkateswaran K . (2010). Diversity of anaerobic microbes in spacecraft assembly clean rooms. Appl Environ Microbiol 76: 2837–2845.
Probst A, Mahnert A, Weber C, Haberer K, Moissl-Eichinger C . (2012). Detecting inactivated endospores in fluorescence microscopy using propidium monoazide. Int J Astrobiol 11: 117–123.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Pruesse E, Peplies J, Glöckner FO . (2012). SINA: accurate high throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28: 1823–1829.
R-project (2011) R Development Core Team R A Language and Environment for Statistical Computing http://www.r-project.org/ . R Foundation for Statistical Computing, Vienna, Austria.
Rawsthorne H, Dock CN, Jaykus LA . (2009). PCR-based method using propidium monoazide to distinguish viable from nonviable Bacillus subtilis spores. Appl Environ Microbiol 75: 2936–2939.
Rogers GB, Stressmann FA, Koller G, Daniels T, Carroll MP, Bruce KD . (2008). Assessing the diagnostic importance of nonviable bacterial cells in respiratory infections. Diagn Microbiol Infect Dis 62: 133–141.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Schloss PD, Gevers D, Westcott SL . (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS one 6: e27310.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR et al. (2006). Microbial diversity in the deep sea and the underexplored "rare biosphere". Proc Natl Acad Sci USA 103: 12115–12120.
Suzuki MT, Taylor LT, DeLong EF . (2000). Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5‘-nuclease assays. App Environ Microbiol 66: 4605–4614.
Tamura K, Dudley J, Nei M, Kumar S . (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599.
Vaishampayan P, Osman S, Andersen G, Venkateswaran K . (2010). High-density 16S microarray and clone library-based microbial community composition of the Phoenix spacecraft assembly clean room. Astrobiology 10: 499–508.
Vaishampayan P, Roberts AH, Augustus A, Schwendner P, Mayilraj S, Salmassi T et al. (2012). Deinococcus phoenicis sp. nov., an extreme ionizing radiation resistant bacterium isolated from the Phoenix Lander assembly facility. Int J System Evol Microbiol 61Pt 6: 1338–1343.
van Frankenhuyzen JK, Trevors JT, Lee H, Flemming CA, Habash MB . (2011). Molecular pathogen detection in biosolids with a focus on quantitative PCR using propidium monoazide for viable cell enumeration. J Microbiol Methods 87: 263–272.
Venkateswaran K, Satomi M, Chung S, Kern RG, Koukol R, Basic C et al. (2001). Molecular microbial diversity of a spacecraft assembly facility. Syst Appl Microbiol 24: 311–320.
Vesper S, McKinstry C, Hartmann C, Neace M, Yoder S, Vesper A . (2008). Quantifying fungal viability in air and water samples using quantitative PCR after treatment with propidium monoazide (PMA). J Microbiol Methods 72: 180–184.
Wagner AO, Malin C, Knapp BA, Illmer P . (2008). Removal of free extracellular DNA from environmental samples by ethidium monoazide and propidium monoazide. Appl Environ Microbiol 74: 2537–2539.
Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG et al. (2012). Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J 6: 94–103.
Yanez MA, Nocker A, Soria-Soria E, Murtula R, Martinez L, Catalan V . (2011). Quantification of viable Legionella pneumophila cells using propidium monoazide combined with quantitative PCR. J Microbiol Methods 85: 124–130.
Acknowledgements
Part of the research described in this study was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under contract with the National Aeronautics and Space Administration. AJP’s contribution was supported by the German National Academic Foundation (Studienstiftung des deutschen Volkes). We are grateful to T DeSantis, L Tom, for PhyloChip analyses, S Westcott and P Schloss, for pyrosequence analysis, J Andy Spry and K Buxbaum for valuable advice and guidance. We thank M Cooper and C Stam for assistance with sample collection and processing, and acknowledge Y Sun at Research and Technology Laboratory for all next-generation sequencing and assistance with TEFAP analyses.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Vaishampayan, P., Probst, A., La Duc, M. et al. New perspectives on viable microbial communities in low-biomass cleanroom environments. ISME J 7, 312–324 (2013). https://doi.org/10.1038/ismej.2012.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2012.114
Keywords
This article is cited by
-
Occurrence of viable but non-culturable (VBNC) pathogenic bacteria in tap water of public places
Frontiers of Environmental Science & Engineering (2024)
-
RNA-based amplicon sequencing is ineffective in measuring metabolic activity in environmental microbial communities
Microbiome (2023)
-
Phylogenomics, phenotypic, and functional traits of five novel (Earth-derived) bacterial species isolated from the International Space Station and their prevalence in metagenomes
Scientific Reports (2023)
-
Metabolic modeling of the International Space Station microbiome reveals key microbial interactions
Microbiome (2022)
-
Microbial Tracking-2, a metagenomics analysis of bacteria and fungi onboard the International Space Station
Microbiome (2022)
