
Abstract
Arbuscular mycorrhizal fungi (AMF) have been symbionts of land plants for at least 450 Myr. It is known that some AMF host in their cytoplasm Gram-positive endobacteria called bacterium-like organisms (BLOs), of unknown phylogenetic origin. In this study, an extensive inventory of 28 cultured AMF, from diverse evolutionary lineages and four continents, indicated that most of the AMF species investigated possess BLOs. Analyzing the 16S ribosomal DNA (rDNA) as a phylogenetic marker revealed that BLO sequences from divergent lineages all clustered in a well-supported monophyletic clade. Unexpectedly, the cell-walled BLOs were shown to likely represent a sister clade of the Mycoplasmatales and Entomoplasmatales, within the Mollicutes, whose members are lacking cell walls and show symbiotic or parasitic lifestyles. Perhaps BLOs maintained the Gram-positive trait whereas the sister groups lost it. The intracellular location of BLOs was revealed by fluorescent in situ hybridization (FISH), and confirmed by pyrosequencing. BLO DNA could only be amplified from AMF spores and not from spore washings. As highly divergent BLO sequences were found within individual fungal spores, amplicon libraries derived from Glomus etunicatum isolates from different geographic regions were pyrosequenced; they revealed distinct sequence compositions in different isolates. Our results show a vertically inherited, monophyletic and globally distributed lineage of endobacteria thriving in AMF cytoplasm. These bacteria split from their sister groups more than 400 Myr ago, colonizing their fungal hosts already before main AMF lineages separated. The BLO–AMF symbiosis can, therefore, be dated back at least to the time when AMF formed the ancestral symbiosis with emergent land plants.
Similar content being viewed by others

Main
Symbiosis is central in the evolution of complexity, being crucial to the lifestyles of animals, plants, fungi and also prokaryotes (Hoffmeister and Martin, 2003). Many functions could only be evolved because of the intimate interaction and often interdependence of different species, as illustrated by endosymbioses, which gave rise to the eukaryotic organelles (Timmis et al., 2004). Scaling up to the ecological level, symbiotically living organisms have key roles in most ecosystems (Moran et al., 2008). Bacterial endosymbionts are widespread in animals, in particular in insects in which they—as ubiquitous and heritable genetic components—offer excellent models to investigate organelle evolution, genome reduction and horizontal gene transfer (Moran et al., 2008). In contrast, examples of endobacteria in fungi are limited to a few examples (Bonfante and Anca, 2009); among them a Burkholderia species living inside a strain of Rhizopus is responsible for fungal pathogenicity, and hence showing the interdependence of the bacterial–fungal alliance (Partida-Martinez and Hertweck, 2005).
Arbuscular mycorrhizal fungi (AMF), which are analyzed in this study, are long known to possess endobacteria in their cytoplasm. AMF belong to the Glomeromycota, a phylum known to be older than the Ascomycota and Basidiomycota (Schüßler et al., 2001), and form symbiotic associations with >80% of land plants, in which both partners benefit from nutrient exchange. This association is commonly described as the result of co-evolution dating back to early Devonian times (Taylor et al., 1995; Redecker et al., 2000; Bonfante and Genre, 2008). As for insect endosymbionts, the presence of endobacteria inside AMF cytoplasm has long been documented by electron microscopy, which has distinguished two bacterial morphotypes. The first, restricted to a phylogenetically relatively young AMF family (Gigasporaceae), is rod shaped, related to Burkholderia and described as an uncultured taxon, Candidatus Glomeribacter gigasporarum (Bianciotto et al., 2003). A fungal line cured from these endobacteria showed that they confer an ecologically relevant fitness to their fungal host (Lumini et al., 2007). The other bacterial type has been detected inside AMF spores and hyphae colonizing plant roots sampled in the field. It is coccoid in shape and has been called ‘bacterium-like organism’ (BLO), as its identity is still obscure (MacDonald et al., 1982; Scannerini and Bonfante, 1991; Schüßler et al., 1994). When BLOs were first detected, knowledge of AMF phylogeny was limited and the fungi were classified only by their morphological features. As a consequence, BLO attribution to specific AMF taxa is uncertain and opens questions of whether BLOs in the fungal cytoplasm represent occasional re-infection by free-living bacteria, or whether they are a consistent feature.
To unambiguously identify the BLOs and assess their distribution in diverse members of the Glomeromycota, we analyzed AMF spores from 28 cultures, representing highly divergent lineages originating from four continents. We used confocal microscopy, fluorescence in situ hybridization (FISH) and electron microscopy to establish BLO localization in the AMF cytoplasm, together with sequencing and phylogenetic analysis on BLOs from single spores or groups. We report that BLOs live only within the fungal cytoplasm and that divergent, but monophyletic bacterial lineages, co-exist in an individual single spore. Unexpectedly, the cell-walled BLOs represent an old, formerly unknown bacterial group that likely falls within the clade harboring the cell wall-lacking Mollicutes. This finding opens new questions about the evolution of Mollicutes-related bacteria, their biotrophic lifestyle, the complexity of interphylum interactions and the symbiotic-genetic makeup of AMF.
Materials and methods
Spore manipulation
AMF cultures were obtained from different culture collections (Supplementary Table S1), in which they had been propagated for several generations either in pot soil cultures in the presence of a plant host or in vitro on root organ cultures (Cranenbrouck et al., 2005). All steps requiring sterility were performed under laminar flow, and plastic material was sterile and DNA free. Spores were cleaned as described in Schwarzott and Schüßler (2001) and surface sterilized with 3% chloramine-T and 0.03% streptomycin.
DNA extraction and amplification
DNA was extracted from spores (either single or groups of five) as described in Lumini et al. (2007) with 10 μl 5 × HotStarTaq PCR buffer (Qiagen, Milan, Italy), or following Schwarzott and Schüßler (2001). The bacterial 16S ribosomal DNA (rDNA) was amplified by PCR with Phusion High-Fidelity DNA polymerase 2 × mastermix (Finnzymes, Espoo, Finland) and the general bacterial primers 16F27 (Bennasar et al., 1996) and 1495r (Bandi et al., 1994) nested with 16F27 and 1387R (Marchesi et al., 1998). The final reaction mix contained 0.02 U μl−1 Phusion polymerase, 1 × Phusion HF Buffer with 1.5 mM MgCl2, 200 μM of each deoxynucleotide triphosphate and 0.5 μM of each primer. Thermal cycling conditions were: 5 min initial denaturation at 99 °C; 30–40 cycles of 10 s denaturation at 98 °C, 30 s annealing at 60 °C and 1 min elongation at 72 °C; and a 10 min final elongation. PCR products were TOPO cloned (Invitrogen, San Giuliano Milanese, Italy) and transformed into Top10 chemically competent Escherichia coli according to the manufacturers instructions. Colonies were screened for insert length by PCR. The PCR products were digested with the restriction endonucleases AluI and RsaI (Invitrogen) (1 U, 1 h at 37 °C) to produce restriction fragment length polymorphism profiles. A modified heat-lysis protocol (Ganguly et al., 2005) was applied to extract plasmids. The plasmid inserts were sequenced from both ends on an ABI 3730 48 capillary sequencer with 50 cm capillary length using BigDye v3.1 sequencing chemistry (Applied Biosystems, Darmstadt, Germany).
Bioinformatic analyses
Sequences were assembled and curated using Seqassem (Sequentix, Klein Raden, Germany) and aligned with the SILVA 16S RNA database (Pruesse et al., 2007), version SSURef_release96, using ARB (Ludwig et al., 2004). Phylogenetic trees were inferred with the PHYLIP package (Felsenstein, 1989), MrBayes (Huelsenbeck and Ronquist, 2001) and RaxML (Stamatakis et al., 2008). Only topologies are shown, which are supported by at least two of the three phylogenetic analysis methods used (neighbor joining, maximum likelihood and Bayesian) with >50% bootstrap or >0.5 posterior probability values. Others are collapsed to polytomies. Dashes instead of numbers indicate that the topology was not supported in the respective analysis (Figure 3). Pairwise distances between sequences types were estimated with ClustalW (Larkin et al., 2007).
Pyrosequencing
A metagenomic approach was adopted for the 454-based experiments; as BLOs are so far uncultured, the starting material was fungal spores. For the construction of amplicon libraries from endobacteria and bacteria associated with the surface of Glomus versiforme Att 475–45, we used three types of samples: (1) washed spores, (2) spores sonicated in 10 μl sterile DNA-free water for 2 min and washed three times afterwards and (3) the water remaining after the sonication. Spores of G. etunicatum culture Att 239–4 were decontaminated as in sample 2. Spores from G. etunicatum isolates MUCL 47650, CA-OT126-3-2 and CA-OT-135-4-2 were collected from in vitro culture and washed twice. Each sample combined five spores or the equivalent water and we used three biological replicates. DNA was extracted as in Schwarzott and Schüßler (2001), and amplified by PCR with Phusion HF Mastermix (as described above) with modified versions of the primers 967F (5′-GCCTCCCTCGCGCCATCAGNNNNCRACGCGNAGAACCTTACC-3′) (Sogin et al., 2006) and 1495r (5′-GCCTTGCCAGCCCGCTCAGCTACGGYTACCTTGTTACGAC-3′) (Bandi et al., 1994), chosen to construct tagged fusion primers (Supplementary Table S3, SI). Thermal cycling conditions were: 5 min initial denaturation at 99 °C; 30 cycles of 10 s denaturation at 98 °C, 30 s annealing at 57 °C and 1 min elongation at 72 °C; and a 10 min final elongation. Each DNA extract was amplified with its respectively tagged fusion primer in five independent PCRs. Products were pooled and purified with the AMPure kit (Agencourt, Beckman Coulter S.p.A, Milan, Italy), quantified with a NanoDrop 1000 spectrophotometer (Thermo Scientific, Euroclone S.p.A., Milan, Italy) and combined equimolar. Product quality was checked on an Agilent 2100 Bioanalyzer (Agilent Technologies, Cernusco, Italy) and sent to BMR Genomic (Padova, Italy) for emulsion PCR and pyrosequencing on a GS FLX sequencer (Roche, Mannheim, Germany).
Bacterial rDNA sequences from G. etunicatum and G. versiforme metagenomic DNA were quality trimmed (quality cutoff set to 20, omitting sequences <150 b), sorted to the respective sample through the tag sequences, and aligned and clustered using the pyrosequencing pipeline provided by the ribosomal database project (Cole et al., 2009). Operational taxonomic units were classified with the ribosomal database project classifier. In addition, a dereplicated subset of sequences (one sequences per sample per operational taxonomic unit recovered at the 3% sequence divergence level) was aligned with SINA (the SILVA web aligner, http://www.arb-silva.de/aligner/; Pruesse et al., 2007), imported and inserted into the ‘All-Species Living Tree’ alignment (Yarza et al., 2008) with ARB. This alignment had been supplemented with the nearly full-length 16S rDNA sequence data of BLOs before use.
Morphological detection of BLOs
The samples were observed with a confocal laser scanning microscope (CLSM, Leica TCS SP2, Leica Microsystems Srl, Milan, Italy) exciting at 488 and 543 nm. A 40 × long-distance water immersion objective (HCX APO N.A. 0.80) and a 63 × water immersion objective (HCX PL APO N.A. 1.20) were used. Images were taken sequentially at each excitation wavelength. Pseudocolors for emission wavelength were green for 500–530 nm (fluorescein isothiocyanate and SYTO BC) and red for 550–580 nm (Cy3) and 605–635 nm (propidium iodide). To rapidly screen AMF spores for endobacteria, they were stained with the dye mixture SYTO BC (1–5 μM; Molecular Probes, San Giuliano Milanese, Italy), known to penetrate intact living bacterial cells. Fungal nuclei and dead bacteria were counterstained with propidium iodide (30–90 μM). Samples were incubated for 15 min at room temperature before spore crushing and observation.
FISH experiments
Before fixation, G. versiforme Att 475–45 spores were vortexed for 2 to 3 min to clean their surface, and washed three times with ultrapure water and once with phosphate-buffered saline. Spores were fixed in 3% formaldehyde buffered with phosphate-buffered saline (Amann et al., 1990) incubated at 4 °C for 3 h or 6 h, washed three times in the saline, suspended in 50% ethanol in the saline, and stored at −20 °C until use.
Oligonucleotide probes were purchased from Sigma-Aldrich (Milan, Italy) and Thermo Fisher Scientific GmbH (Ulm, Germany), 5′-end labeled with Cy3 or fluorescein isothiocyanate. The eubacterial probe EUB338I (Amann et al., 1990) was used as a general probe, and Apis2Pa (Moran et al., 2005), which targets the genus Buchnera, was used as a negative control to detect nonspecific binding. Specific oligonucleotide probes were designed with ARB. They matched bacterial sequences amplified from Glomus group B (Glomerales) and Diversisporales and showed a minimum of three centrally located mismatches to all other sequences in the ARB 16S database. The specificity of BLOgrBC (5′-GCCAATCCTACCCTTGTCA-3′) was tested empirically. Fungal spores were immobilized on 10-well microscope slides (6 mm Ø each well, Structure Probe, West Chester, PA, USA) with a drop of 0.5% agarose (Sigma-Aldrich) and dehydrated in 50%, 75% and 100% ethanol. Spores were crushed to allow penetration of the probes into the cytoplasm during hybridization, as described in Bertaux et al. (2005). The probes were hybridized at a stringency of 35% formamide (Sigma-Aldrich).
Microscope slides were mounted with SlowFade Antifade kit component A (Molecular Probes). The hybridization to EUB338I and BLOgrBC was repeated 14 times, corresponding to a total of 40 G. versiforme spores.
Transmission electron microscopy
To allow fixative penetration into the spore, spore walls of surface-sterilized G. versiforme Att 475–45 spores were incised with a fine syringe needle (U100 Insulin ACCU-FINE; Roche, Monza, Italy). These spores were immediately transferred to 50 mM sodium cacodylate buffer (pH 7) containing in 2.5% (v/v) glutaraldehyde and then processed as described in Bianciotto et al. (2003) and Lumini et al. (2007).
Ultrathin sections (70 nm) were cut with an ultramicrotome (Ultracut; Reichert and Jung, Vienna, Austria) counterstained with uranyl acetate and lead citrate and observed with a transmission electron microscope (Philips CM10, Philips Medical Systems, Eindhoven, The Netherlands). In all, 10 individual spores were examined.
Results
BLOs are present in diverse AMF families
Spores from 28 cultures, including in vitro-grown root organ cultures and pot cultures, were studied. They represent 16 phylogenetically characterized species belonging to diverse Glomeromycota lineages and originating from four continents (Figure 1; Supplementary Table S1, SI). After staining with the fluorescent dyes SYTO BC and propidium iodide, the BLOs were observed within the spore cytoplasm as green fluorescent spots approximately 500 nm in diameter, whereas fungal nuclei appeared red (Figure 2a). BLOs were consistently found in 19 of the cultures and were associated with 11 of the 16 AMF species tested, being present in members of the Ambisporaceae and Geosiphonaceae (Archaeosporales, an ancient AMF lineage), Glomus group A and Glomus group B (corresponding to two families in the Glomerales), and Diversisporaceae, Gigasporaceae and Acaulosporaceae (Diversisporales) (Figure 1; Supplementary Table S1, SI).

Scheme of taxonomic relationships between AMF isolates and cultures used for molecular and cell biological investigations. The order level is indicated: Archaeosporales (1), Diversisporales (2) and Glomerales (3). Glomerales can be further split into the Glomus group Aa, Ab and B. The fungal species is followed by the respective culture name, and * (isolates in pot culture), + (isolates in vitro on root organ culture (ROC)) or ∼ (culture in vitro on whole plants (WPC)), and other cultures are multispore cultures (not isolates). The geographical origins are in Asia (AS), Europe (EU), North America (NA), Central America (CA) and South America (SA).

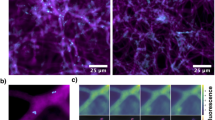
Bacterial structures inside AMF. (a) Bacterial and fungal structures in the fungal cytoplasm (FC) of a manually crushed G. etunicatum spore, stained with the fluorescent dyes SYTO BC and propidium iodide (PI). Inset: enlarged portion of cytoplasm. Living bacteria (LB) appear as coccoid structures in green, whereas fungal nuclei (FN), dead bacteria and the fungal spore wall (FW) appear in red. Scale bars, 10 μm. (b) Electron micrograph of a BLO inside G. versiforme cytoplasm, showing a prominent Gram-positive cell wall (BW). Scale bars, 0.5 μm. The color reproduction of this figure is available on the html full text version of the manuscript.
BLOs are related to the Mollicutes
Bacterial 16S rDNA was amplified from surface-sterilized or in vitro-cultured AMF spores using general bacterial primers. A set of BLO-specific primers was then developed, based on the sequence data obtained. In total, 107 distinct sequence types were obtained from 17 of the 28 cultures, including members of the Glomerales (G. caledonium, G. mosseae, G. claroideum and G. etunicatum), Diversisporales (Gigaspora margarita, Scutellospora gilmorei and G. versiforme) and the Archaeosporales (Ambispora appendicula, A. fennica and Geosiphon pyriformis). BLO 16S rDNA could not be amplified from an Acaulospora sp. and Kuklospora colombiana (Acaulosporaceae, Diversisporales), notwithstanding the morphological evidence of bacterial structures by fluorescent staining.
All sequences clustered in a well-supported monophyletic clade that was not closely related to any described bacterial group. BLOs therefore represent a new, undescribed bacterial higher taxon that, although there is a distinct cell wall (Figure 2b), forms a subclade within the wall-less Mollicutes (Ludwig et al., 2008) (Figure 3) likely being a sister clade to the Entomoplasmatales and Mycoplasmatales.

Phylogenetic tree based on the 16S rDNA, showing the position of BLO sequences within the clades of Mollicutes (red) and Firmicutes (gray); Cyanobacteria (dark green) sequences are used as outgroup. BLO sequences are grouped and coded in different colors according to the associated AMF species. Shades of the same color are applied to sequences from the same AMF family. One to six subclades of BLO sequences were retrieved per AMF species. Where the branching pattern is resolved, BLO sequences from closely related AMF are found to group together. Support values given are from maximum likelihood/neighbor joining/Bayesian analyses. Dashes instead of numbers imply that the topology was not supported in the respective analysis. The organization within the clades is shown in Supplementary Figure S1 a and b (SI).
Highly divergent BLO sequences exist within a single AMF spore
Within the monophyletic BLO clade (Figure 3), highly variable 16S rDNA BLO sequences were found, showing up to 20% intrasporal sequence divergence even within single AMF spores. BLO sequences from an AMF species formed from one (A. fennica) up to six divergent subclades (G. claroideum). The branches in the basal part of the tree included sequence types from the Archaeosporales, Diversisporales and Glomerales, and their topological relationships could not be resolved. In contrast, the more terminal part of the tree was better resolved. It only contained bacterial sequences from Glomerales and Diversisporales (Supplementary Figure S1, SI). Here, BLO sequences stemming from closely related fungi clustered together, indicating co-evolution with their hosts.
BLOs are located within the AMF cytoplasm
To unambiguously show that BLOs are located in the cytoplasm of glomeromycotan fungi, G. versiforme (Att 475–45) was selected because of easy production and handling of spores, low degree of cytoplasm autofluorescence and its long-standing use as a model fungus in arbuscular mycorrhizal research (e.g., Harrison and Buuren, 1995). The 16S rDNA sequences obtained in our work allowed us to develop specific probes for FISH. The new BLO-specific probe BLOgrBC colocalized with the widely used general bacterial probe EUB338I within the G. versiforme cytoplasm (Figure 4) in which it labeled coccoid structures (Figure 4, compare with Figure 2a). The FISH signal was not detected in any free-living bacteria or those associated with the spore surface (data not shown). FISH also indicated that the structures stained by SYTO BC were indeed bacteria.

FISH on a crushed G. versiforme spore. The location of the spore wall (FW) and the squeezed out cytoplasm (FC) is shown in transmitted light (left side). Bacteria are seen as coccoid fluorescent spots in the fungal cytoplasm (indicated by arrows), their 16S rRNA hybridized to the BLO-specific probe BLOgrBC (red) and to the general eubacterial probe EUB338I (green). Both probes label the same structures, as seen in the superimposition of the signals (right side). The insets show part of the cytoplasm enlarged. Scale bars, 10 μm.
Electron microscopy of the same G. versiforme culture confirmed the presence of coccoid bacteria in the fungal cytoplasm. They were 250–500 nm in size, possessed a homogeneous, Gram-positive type cell wall and, unlike Candidatus Glomeribacter, were not surrounded by a fungal membrane (Figure 2b).
To confirm the cytoplasmic localization of the BLOs we used the 454 GS-FLX technology (Margulies et al., 2005) to sequence 16S rDNA amplicon libraries. The metagenomic DNA was derived from either (1) five washed spores, (2) five sonicated and washed spores or (3) the water in which the five spores were held during sonication, with three biological replicates each. A total of 1125 high-quality sequences were obtained. Sequences detected from the sonicated water were considered as derived from spore surface-associated bacteria. The results showed that sonication did not remove all surface bacteria but the percentage of non-BLO sequences associated with the surface of sonicated spores was lower than for spores only washed (Figure 5; Supplementary Figure S2, SI). They belonged to bacterial groups already reported as associated with AMF spores (reviewed in Bonfante and Anca, 2009) and in studies of soil biodiversity (Roesch et al., 2007; Elshahed et al., 2008; Kielak et al., 2008). In contrast, BLO sequences were exclusively obtained from washed and sonicated spores but never from sonicated water. This points to their presence inside fungal spores and more importantly shows that BLOs were not detected as free-living bacteria. Sequences related to those of BLOs have been reported in some recent studies. With the exception of one that was shown to be associated with Gigaspora margarita (Long et al., 2009), all the other sequences are annotated as environmental and have been retrieved in studies on soil bacterial diversity, from tree and grassland environments (Dunbar et al., 2002; Elshahed et al., 2008; Hansel et al., 2008; Lesaulnier et al., 2008; Cruz-Martinez et al., 2009). Most likely, these sequences originated from BLO-harboring AMF present in the samples, either as spores or, hyphae in the soil or in root fragments.

Sequences recovered from 16S rDNA amplicon libraries of G. versiforme are classified as BLOs (gray) or non-BLOs (black); the percentage of sequences that can be attributed to either group is shown. The metagenomic DNA was derived from washed spores (W), sonicated and washed spores (S) or the water in which the spores were sonicated (H), with three biological replicates each (1, 2 and 3). Spores were checked and remained intact after sonication; therefore, bacterial sequences retrieved from the sonicated water were considered as originating from surface-associated bacteria. Sonication did not remove all surface bacteria, but the percentage of non-BLO sequences associated with the surface of sonicated spores was lower than for spores only washed, and no BLO sequences were recovered from (H).
BLO rDNA sequence variability: another source of genetic variability in arbuscular mycorrhizal fungi
The model AMF Glomus sp. ‘intraradices-like’ DAOM197198, whose genome is currently under sequencing, has been indicated to contain multiple, polymorphic genomes (Hijri and Sanders, 2005; Martin et al., 2008). In contrast, the mitochondrial genome of this fungus and its close relatives (Lee and Young, 2009) including the mt-rDNA (Börstler et al., 2008), is nearly invariable. For isolates of G. intraradices and the species represented by Glomus sp. DAOM197198, exceptionally variable nuclear rDNA regions were reported with up to >20% intrasporal sequence divergence in the internal transcribed spacer region (Stockinger et al., 2009). A similarly high internal transcribed spacer variability was recently also reported for AMF from other phylogenetic clades (Stockinger et al., 2010). Unexpectedly, a very high variability (up to 20% intrasporal sequence divergence) was found between BLO 16S rDNA sequences attributed to a single fungal culture and even to single spores in the present study, although not all these variations could be recovered from each culture. To understand whether this was due to insufficient sampling density, amplicon libraries of G. etunicatum spores were 454 GS-FLX pyrosequenced.
G. etunicatum was chosen because four cultures were available for comparison. Three of these were single spore isolates established as root organ cultures (CA-OT-126-3-2, CA-OT-135-4-2 and MUCL47650), and one was propagated in pot (Att 239–4) (Figure 1; Supplementary Table S1, SI). Each library was again generated from five spores, with three biological replicates each, leading to 2827 sequences. To define phylotypes, all sequences with ⩽3% divergence were clustered. Only one of the resulting 20 BLO phylotypes was present in all four G. etunicatum cultures, one was exclusively detected in the biological replicates of MUCL47650 and Att 239–4, one only in the biological replicates of CA-OT-135-4-2, whereas several other phylotypes could be attributed to both CA-OT-126-3-2 and CA-OT-135-4-2 isolates, both originating from California (Table S2, SI). Thus, pyrosequencing confirmed that different phylotype compositions do indeed exist between isolates of the same AMF species, and highlighted that the BLO sequences represent the vast majority (89.8%) of the bacterial sequences recovered from cleaned spores. When the in vitro cultures, expected to be free of contaminating bacteria, were considered, BLO sequences represented even 93.6%. Although the BLO phylotypes were present throughout all biological replicates of a sample, the remaining non-BLO sequences were highly diverse and variable in between replicates, indicating contaminants acquired during sample preparation. However, the percentage values have to be interpreted with care, as sequences may be artificially replicated during pyrosequencing (Gomez-Alvarez et al., 2009).
Discussion
Mycorrhizas, the symbioses established by most land plants and soil fungi, are often described as the result of tripartite interactions, as many bacteria are known to be associated as free-living helper microbes (Frey-Klett et al., 2007). In contrast, existence of true endobacteria in mycorrhizal fungi has been convincingly shown only for the betaproteobacterium Candidatus Glomeribacter gigasporarum, whose presence is restricted to members of the family Gigasporaceae (Bonfante and Anca, 2009). In this study we show that the Gram positive endobacteria (the BLOs) live in the cytoplasm of many AMF lineages, and that their occurrence is not a sporadic event. BLOs are present in Glomus group A and B of the Glomerales, in Diversisporales and in Archeosporales, covering diverse evolutionary lineages in the AMF. Interestingly, they were not detected in Glomus sp. ‘intraradices-like’ DAOM197198, G. intraradices and G. clarum, which are all closely related and belong to the subclade Glomus group Ab (Schwarzott et al., 2001).
The AMF-BLO sequences cluster within those of the Mollicutes, despite the presence of a distinct cell wall and represent a new bacterial higher taxon. The AMF cultures that tested positive for BLOs originated from four continents and represent diverse families, including more basal evolutionary lineages. This indicates that BLOs already colonized AMF before the split of those early diverging AMF lineages. As a consequence, and from the phylogenetic tree topology, the BLOs must have been living in AMF for more than 400 Myr. Because many of the AMF cultures studied here were maintained in in vitro root organ cultures, we also obtained evidence that BLOs are vertically transmitted, similar to many endobacteria living in insects (Baumann & Moran, 1997) and Candidatus Glomeribacter gigasporarum inside AMF of the Gigasporaceae.
Theory predicts that vertically transmitted symbionts, for example, in insects, should offer some benefit to the host as they are maintained by it (Brownlie and Johnson, 2009). Even if, at the moment, we have no clue to BLO function, we can infer that BLOs likely confer fitness on their fungal hosts. Electron microscopy shows that BLOs are directly embedded in the fungal cytoplasm, without a surrounding host membrane, indicating a high level of compatibility or an ancient interaction.
Taken together, our results show that AMF-BLOs are pan-global, vertically inherited, monophyletic, ancient and, so far, uncultured and most likely Mollicutes-related endobacteria. Although the 16S phylogenies place BLOs with Mollicutes rather than with Firmicutes, the exact placement of the BLO clade should currently be treated with caution. The relationship between Mollicutes, which belong to the Tenericutes, and the Firmicutes is in fact not clearly solved (Ludwig et al., 2008; Battistuzzi and Hedges, 2009). To unambiguously resolve the relationships within these phyla and then attribute BLOs to one of them, data from other genes will be required. Nevertheless, the finding that the BLOs, Entomoplasmatales and Mycoplasmatales likely share a common ancestor raises questions about the biology and evolution of the Mollicutes. A common characteristic is their endosymbiotic or parasitic lifestyle (Johansson and Pettersson, 2002). The intracellular status of the AMF-BLOs supports the concept that the ancestors of Mollicutes had already evolved mechanisms to exploit intracellular or symbiotic niches, as showed by the present-day species in the genera Phytoplasma and Mycoplasma, which are widespread parasites of animals and plants. Intriguingly, the BLO cell wall, absent from other members of the Mollicutes, indicates that BLOs maintained a trait that sister clades have lost, after the divergence of Mollicutes from their Firmicute-related ancestors, between 600 and 2000 Myr (Maniloff, 2002; Battistuzzi and Hedges, 2009). The presence of a cell wall and the phylogenetic branching of the BLOs support the concept of an ancestral association between BLOs and AMF. Because AMF themselves have been involved in symbioses with plants since Devonian times (Taylor et al., 1995; Redecker et al., 2000), our findings open new questions on the complexity and age of multiple and symbiotic inter-phylum interactions.
Lastly, in the broader context of eukaryotic cell evolution, the plant-symbiont AMF have been unique up to now; they maintain an extraordinarily high polymorphism in their genomes (Martin et al., 2008), as reflected by intrasporal nuclear rDNA variability (Stockinger et al., 2009, 2010), likely resulting in a population of heterokaryotic nuclei. The variability of BLO rDNA sequences now indicates a similar variability for the AMF-associated endobacterial population, even within one fungal spore. BLOs seem to be widespread not only within the AMF maintained in culture, as investigated in this study, but also in environmental samples, as is emerging from ultrastructural observations (e.g., Ligrone et al., 2007). For this reason, their pan-global distribution and genetic variability, apparently associated with avoidance of a bottleneck, add a further level of interest to the still enigmatic AMF genomes.
References
Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA . (1990). Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56: 1919–1925.
Bandi C, Damiani G, Magrassi L, Grigolo A, Fani R, Sacchi L . (1994). Flavobacteria as intracellular symbionts in cockroaches. Proc Biol Sci 257: 43–48.
Battistuzzi FU, Hedges SB . (2009). A major clade of prokaryotes with ancient adaptations to life on land. Mol Biol Evol 26: 335–343.
Baumann P, Moran NA . (1997). Non-cultivable microorganisms from symbiotic associations of insects and other hosts. Antonie van Leeuwenhoek 72: 39–48.
Bennasar A, Rosselló-Mora R, Lalucat J, Moore ER . (1996). 16S rRNA gene sequence analysis relative to genomovars of Pseudomonas stutzeri and proposal of Pseudomonas balearica sp. nov. Int J Syst Bacteriol 46: 200–205.
Bertaux J, Schmid M, Hutzler P, Hartmann A, Garbaye J, Frey-Klett P . (2005). Occurrence and distribution of endobacteria in the plant-associated mycelium of the ectomycorrhizal fungus Laccaria bicolor S238N. Environ Microbiol 7: 1786–1795.
Bianciotto V, Lumini E, Bonfante P, Vandamme P . (2003). ‘Candidatus Glomeribacter gigasporarum’ gen nov, sp nov, an endosymbiont of arbuscular mycorrhizal fungi. Int J Syst Evol Microbiol 53: 121–124.
Börstler B, Raab PA, Thiéry O, Morton JB, Redecker D . (2008). Genetic diversity of the arbuscular mycorrhizal fungus Glomus intraradices as determined by mitochondrial large subunit rRNA gene sequences is considerably higher than previously expected. New Phytol 180: 452–465.
Bonfante P, Anca IA . (2009). Plants, mycorrhizal fungi, and bacteria: a network of interactions. Annu Rev Microbio 63: 363–383.
Bonfante P, Genre A . (2008). Plants and arbuscular mycorrhizal fungi: an evolutionary-developmental perspective. Trends Plant Sci 13: 492–498.
Brownlie JC, Johnson KN . (2009). Symbiont-mediated protection in insect hosts. Trends Microbiol 17: 348–354.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucl Acids Res 37: D141–D145.
Cranenbrouck S, Voets L, Bivort C, Renard L, Strullu DG, Declerck S . (2005). Methodologies for in vitro cultivation of arbuscular mycorrhizal fungi with root organs. In: Declerck S, Strullu D, Fortin JA (eds). In Vitro Culture of Mycorrhizas. Springer: Heidelberg, pp 341–375.
Cruz-Martinez K, Suttle KB, Brodie EL, Power ME, Andersen GL, Banfield JF . (2009). Despite strong seasonal responses, soil microbial consortia are more resilient to long-term changes in rainfall than overlying grassland. ISMEJ 3: 738–744.
Dunbar J, Barns SM, Ticknor LO, Kuske CR . (2002). Empirical and theoretical bacterial diversity in four Arizona soils. Appl Environ Microbiol 68: 3035–3045.
Elshahed MS, Youssef NH, Spain AM, Sheik C, Najar FZ, Sukharnikov LO et al. (2008). Novelty and uniqueness patterns of rare members of the soil biosphere. Appl Environ Microbiol 74: 5422–5428.
Felsenstein J . (1989). PHYLIP-phylogeny inference package (version 3.2). Cladistics 5: 164–166.
Frey-Klett P, Garbaye J, Tarkka M . (2007). The mycorrhiza helper bacteria revisited. New Phytol 176: 22–36.
Ganguly T, Chen P, Teetsel R, Zhang LP, Papaioannou E, Cianciarulo J . (2005). High-throughput sequencing of high copy number plasmids from bacterial cultures by heat lysis. BioTechniques 39: 304 306, 308.
Gomez-Alvarez V, Teal TK, Schmidt TM . (2009). Systematic artifacts in metagenomes from complex microbial communities. ISMEJ 3: 1314–1317.
Hansel CM, Fendorf S, Jardine PM, Francis CA . (2008). Changes in bacterial and archaeal community structure and functional diversity along a geochemically variable soil profile. Appl Environ Microbiol 74: 1620–1633.
Harrison MJ, Buuren MLV . (1995). A phosphate transporter from the mycorrhizal fungus Glomus versiforme. Nature 378: 626–629.
Hijri M, Sanders IR . (2005). Low gene copy number shows that arbuscular mycorrhizal fungi inherit genetically different nuclei. Nature 433: 160–163.
Hoffmeister M, Martin W . (2003). Interspecific evolution: microbial symbiosis, endosymbiosis and gene transfer. Environ Microbiol 5: 641–649.
Huelsenbeck JP, Ronquist F . (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755.
Johansson K, Pettersson B . (2002). Taxonomy of mollicutes. In: Razin S, Herrmann R (eds). Molecular Biology and Pathogenicity of Mycoplasmas. Springer: Heidelberg, pp 1–30.
Kielak A, Pijl AS, Veen JAV, Kowalchuk GA . (2008). Differences in vegetation composition and plant species identity lead to only minor changes in soil-borne microbial communities in a former arable field. FEMS Microbiol Ecol 63: 372–382.
Larkin M, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948.
Lee J, Young JPW . (2009). The mitochondrial genome sequence of the arbuscular mycorrhizal fungus Glomus intraradices isolate 494 and implications for the phylogenetic placement of Glomus. New Phytol 183: 200–211.
Lesaulnier C, Papamichail D, McCorkle S, Ollivier B, Skiena S, Taghavi S et al. (2008). Elevated atmospheric CO2 affects soil microbial diversity associated with trembling aspen. Environ Microbiol 10: 926–941.
Ligrone R, Carafa A, Lumini E, Bianciotto V, Bonfante P, Duckett J . (2007). Glomeromycotean associations in liverworts: a molecular, cellular, and taxonomic analysis. Am J Bot 94: 1756–1777.
Long L, Yao Q, Ai Y, Deng M, Zhu H . (2009). Detection of a novel bacterium associated with spores of the arbuscular mycorrhizal fungus Gigaspora margarita. Can J Microbiol 55: 771–775.
Ludwig W, Schleifer K, Whitman WB . (2008). Revised road map to the phylum Firmicutes. In: De Vos P, Garrity GM, Jones D, Rainey FA, Schleifer KH, Whitman WB (eds). Bergey's Manual of Systematic Bacteriology. Springer: Heidelberg.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar AB et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Lumini E, Bianciotto V, Jargeat P, Novero M, Salvioli A, Faccio A et al. (2007). Presymbiotic growth and sporal morphology are affected in the arbuscular mycorrhizal fungus Gigaspora margarita cured of its endobacteria. Cell Microbiol 9: 1716–1729.
MacDonald RM, Chandler M, Mosse B . (1982). The occurrence of bacterium-like organelles in vesicular-arbuscular mycorrhizal fungi. New Phytol 90: 659–663.
Maniloff J . (2002). Phylogeny and evolution. In: Razin S, Herrmann R (eds). Molecular Biology and Pathogenicity of Mycoplasmas. Springer: Heidelberg, pp 31–44.
Marchesi JR, Sato T, Weightman AJ, Martin TA, Fry JC, Hiom SJ et al. (1998). Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl Environ Microbiol 64: 2333.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380.
Martin F, Gianinazzi-Pearson V, Hijri M, Lammers P, Requena N, Sanders IR et al. (2008). The long hard road to a completed ‘Glomus intraradices’ genome. New Phytol 180: 747–750.
Moran NA, McCutcheon JP, Nakabachi A . (2008). Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42: 165–190.
Moran NA, Russell JA, Koga R, Fukatsu T . (2005). Evolutionary relationships of three new species of Enterobacteriaceae living as symbionts of aphids and other insects. Appl Environ Microbiol 71: 3302–3310.
Partida-Martinez LP, Hertweck C . (2005). Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature 437: 884–888.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Redecker D, Kodner R, Graham LE . (2000). Glomalean fungi from the Ordovician. Science 289: 1920–1921.
Roesch LFW, Fulthorpe RR, Riva AR, Casella G, Hadwin AKM, Kent AD et al. (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISMEJ 1: 283–290.
Scannerini S, Bonfante P . (1991). Bacteria and bacteria-like objects in endomycorrhizal fungi. In: Margulis L, Fester R (eds). Symbiosis as a Source of Evolutionary Innovation: Speciation and Morphogenesis. MIT Press: Cambridge, MA, pp 273–287.
Schüßler A, Mollenhauer D, Schnepf E, Kluge M . (1994). Geosiphon pyriformis an endosymbiotic association of fungus and cyanobacteria: the spore structure resembles that of arbuscular mycorrhizal (AM) fungi. Botanica Acta 107: 36–45.
Schüßler A, Schwarzott D, Walker C . (2001). A new fungal phylum, the Glomeromycota: phylogeny and evolution. Mycol Res 105: 1413–1421.
Schwarzott D, Schüßler A . (2001). A simple and reliable method for SSU rRNA gene DNA extraction, amplification, and cloning from single AM fungal spores. Mycorrhiza 10: 203–207.
Schwarzott D, Walker C, Schüßler A . (2001). Glomus, the largest genus of the arbuscular mycorrhizal fungi (Glomales), is nonmonophyletic. Mol Phylogenet Evol 21: 190–197.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR et al. (2006). Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA 103: 12115–12120.
Stamatakis A, Hoover P, Rougemont J . (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57: 758–771.
Stockinger H, Walker C, Schüßler A . (2009). Glomus intraradices DAOM197198 a model fungus in arbuscular mycorrhiza research, is not Glomus intraradices. New Phytol 183: 1176–1187.
Stockinger H, Krüger M, Schüßler A . (2010) DNA barcoding of arbuscular mycorrhizal fungi. New Phytol, in press.
Taylor TN, Remy W, Hass H, Kerp H . (1995). Fossil arbuscular mycorrhizae from the early Devonian. Mycologia 87: 560–573.
Timmis JN, Ayliffe MA, Huang CY, Martin W . (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat Rev Genet 5: 123–135.
Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer KH et al. (2008). The All-Species Living Tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol 31: 241–250.
Acknowledgements
We thank T Pawlowska (USA), C Walker (GB), the Genebank at NIAS (Japan), S Cranenbrouck and S Declerck from GINCO (Belgium), as well as all original collectors for providing AMF cultures. We thank V Bianciotto and E Lumini for useful comments and A Faccio for TEM preparation. The research leading to these results received funding from the European Community's Sixth Framework Programme (FP6/2005–2009) under grant agreement no. MEST-CT-2005-021016 (TRACEAM), from Compagnia di San Paolo, Torino and from Converging technology Project (BIOBIT, CIPE) to PB.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Naumann, M., Schüßler, A. & Bonfante, P. The obligate endobacteria of arbuscular mycorrhizal fungi are ancient heritable components related to the Mollicutes. ISME J 4, 862–871 (2010). https://doi.org/10.1038/ismej.2010.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.21
Keywords
This article is cited by
-
Characterization of microbial communities during Grifola frondosa (maitake) wood log cultivation
Journal of Wood Science (2023)
-
Coniochaeta fungus benefits from its intracellular bacteria to form biofilm and defend against other fungi
Archives of Microbiology (2021)
-
Morphological diversity of fungal reproductive units in the Lower Devonian Rhynie and Windyfield cherts, Scotland: a new species of the genus Windipila
PalZ (2020)
-
Microbial interactions within the plant holobiont
Microbiome (2018)
