
Abstract
The inhibition of aldosterone activity is a useful approach for preventing the progression of cardiovascular and renal diseases in hypertensive patients. Although the results of our previous in vivo study suggested that N-type calcium channels may have a role in regulating plasma aldosterone levels, the direct relationship between N-type calcium channels and aldosterone production in adrenocortical cells has not been examined. In this study, the analysis of quantitative reverse transcription-PCR, western blotting, and immunocytological staining indicated the possible presence of N-type calcium channels in human adrenocortical cells (H295R cell line). Patch clamp analysis indicated that omega-conotoxin GVIA (CnTX), an N-type calcium channel inhibitor, suppressed voltage-dependent barium currents. During steroidogenesis, CnTX significantly reduced the transient calcium signaling induced by angiotensin II (Ang II) and partially prevented Ang II-induced aldosterone and cortisol formation with no significant influence on CYP11B2 and CYP11B1 mRNA expression. In addition, in α1B calcium channel subunits, knockdown significantly decreased Ang II-induced aldosterone formation with increments in CYP11B2 mRNA expression. We also investigated the inhibitory activities of some types of dihydropyridine calcium channel blockers (CCBs; cilnidipine: L-/N-type CCB, efonidipine: L-/T-type CCB, and nifedipine: L-type CCB), and these agents showed a dose-dependent inhibition effect on Ang II-induced aldosterone and cortisol production. Furthermore, only cilnidipine failed to suppress CYP11B1 expression in H295R cells. These results suggest that N-type calcium channels have a significant role in transducing the Ang II signal for aldosterone (and cortisol) biosynthesis, which may explain the mechanism by which N-type calcium channels regulate plasma aldosterone levels.
Similar content being viewed by others


Introduction
Aldosterone is considered a key regulator of fluid and electrolyte balance in mammals,1 and it has a central role in the pathogenesis of chronic heart failure and chronic kidney disease.2, 3 Indeed, in patients with heart failure or hypertension, aldosterone synthesis is activated in both the adrenal gland and the heart.4, 5 Aldosterone secretion from the adrenal gland is diminished by the administration of a sodium-based diet or by controlling the levels of potassium, angiotensin II (Ang II), or adrenocorticotropic hormone.6 In human adrenal zona glomerulosa cells, Ang II has a primary role in the regulation of aldosterone biosynthesis, secretion, and cell growth, and upregulates numerous genes.7 Ang II-stimulated cortisol and aldosterone production involves a depolarization of the plasma membrane8 and the activation of calcium influx through voltage-gated channels.9 Although the regulation of Ang II-induced aldosterone production is well established, the modulating factors affecting this regulation or its downstream signaling components, such as voltage-gated calcium channels, have not been completely elucidated.
Calcium channel blockers (CCBs) are commonly used to treat hypertension and generally act on L-type calcium channels. Recent studies have shown that some CCBs can also block channels belonging to other subtypes, such as N-type and T-type channels. The blockade of non-L-type calcium channels exerts unique effects. Recent studies have revealed that adrenal glomerulosa cells express both L-type and T-type calcium channels. Furthermore, T-type calcium channels, not the L-type calcium channels, have an essential role in aldosterone production.10 Meanwhile, previous studies have revealed that cilnidipine, an N-type and L-type CCB, is capable of blocking the two types of channels.11
N-type calcium channels are members of the major calcium channel family, but they are fundamentally different from the other calcium channels and have highly distinctive calcium-regulation mechanisms.12, 13 N-type calcium channels are heteromultimeric complexes consisting of a pore-forming α1 subunit (α1B) and at least two auxiliary subunits, β and α2δ, which modulate the function of the α1 subunit.14 N-type calcium channels have been identified in several types of cells,15, 16, 17 and these channels have been reported to mediate rapid calcium influx into the synaptic terminal, which triggers synaptic vesicle exocytosis, neurotransmitter release, synaptogenesis, and gene expression.18, 19, 20
We have reported that cilnidipine reduced the plasma aldosterone level in spontaneously hypertensive rats21 and in a canine model of chronic atrioventricular block that is known to show ventricular electrical remodeling;22 however, the L-type CCB, amlodipine, did not show any suppressive effect on plasma aldosterone levels. These results suggest that N-type calcium channels are important regulators of the plasma aldosterone level. N-type calcium channels have a critical role in gating transmitter release from synaptic nerve terminals; however, little is known regarding the other functions of these channels, including their roles in steroidogenesis in adrenal glomerulosa cells.
Therefore, in this study, we aimed to determine the presence and functional roles of the N-type calcium channels in the adrenal cortex, which is a major source of aldosterone. To this end, we used a human adrenocarcinoma (H295R) cell line,23 and the depolarizing response of N-type calcium channels was confirmed by patch clamp techniques. In addition, the steroidogenesis response of N-type calcium channels was examined by using omega-conotoxin GVIA (CnTX; an N-type calcium channel inhibitor) and other CCBs such as cilnidipine (L-type/N-type CCB), nifedipine (L-type CCB), and efonidipine (L-type/T-type CCB). This response was analyzed using Ang II to determine whether N-type calcium channels regulate Ang II-induced aldosterone secretion and contribute to Ang II-induced organ damage.
Methods
Reagents
Cilnidipine was synthesized at Ajinomoto Pharmaceuticals (Kawasaki, Kanagawa, Japan). Efonidipine was extracted from tablets in our laboratories. Nifedipine was purchased from Sigma-Aldrich (St Louis, MO, USA), and Ang II and CnTX were purchased from the Peptide Institute (Osaka, Japan). Each calcium channel inhibitor was initially dissolved in dimethyl sulfoxide (final concentration, 0.1%; dimethyl sulfoxide; Sigma-Aldrich), and Ang II was dissolved in Dulbecco's modified essential medium/F12 medium (Invitrogen, Carlsbad, CA, USA) containing 0.2% Nu-serum (BD Biosciences, Bedford, MA, USA).
Cell culture
The NCI-H295R human adrenocortical cell line (H295R) was obtained from the American Type Culture Collection and cultured in Dulbecco's modified essential medium/F12 medium containing 2% Nu-serum, 1% ITS plus (BD Biosciences), and antibiotics (penicillin and streptomycin; Sigma-Aldrich). The cells were maintained at 37 °C in a humidified atmosphere containing air and carbon dioxide (95%/5%, vol/vol).24
Assay for steroid production
H295R cells were cultured in 12-well plates until 80% confluence. Next, the medium was replaced with fresh medium containing 0.2% Nu-serum, and the cells were incubated for an additional 24 h. Then, the culture supernatants were replaced with medium containing Ang II, including vehicle (0.1% dimethyl sulfoxide) or each calcium channel inhibitor. After 48 h of incubation, the steroid content in the conditioned media and the cellular protein content were determined.
The aldosterone and cortisol secreted into the H295R culture medium were measured using the Aldosterone Express EIA Kit (Cayman Chemical, Ann Arbor, MI, USA) and the Cortisol Express EIA Kit (Cayman Chemical), respectively. All steroid levels were normalized to the cellular protein content per well, which was determined by performing the bicinchoninic acid protein assay using a bicinchoninic acid assay kit (Pierce Chemical, Rockford, IL, USA).25
Current recordings
Whole-cell mode of the patch clamp technique was carried out at 22–25 °C with EPC 9 (HEKA Elektronik, Pfalz, Germany) or Axopatch 200B (Molecular Devices, Silicon Valley, CA, USA) patch clamp amplifiers as previously described.26 Patch pipettes were made from borosilicate glass capillaries (1.5 mm outer diameter, 0.87 mm inner diameter; Hilgenberg, Malsfeld, Germany) using a P-97 Flaming/Brown micropipette puller (Sutter Instrument, Novato, CA, USA). Pipette resistance ranged from 2.5 to 3.5 MΩ when filled with the pipette solutions as described below. The series resistance was electronically compensated to >60%, and both the leakage and the remaining capacitance were subtracted by the −P/4 method. Currents were sampled at 100 kHz after low pass filtering at 8.4 kHz (3 db) in experiments of activation kinetics or otherwise sampled at 20 kHz after low pass filtering at 3.0 kHz (3 db). Data were collected and analyzed using the Pulse (version 8.77, HEKA Elektronik) or the Clampex programs (version 10.2, Molecular Devices). The external solution contained (in mM) 10 BaCl2, 143 tetraethylammonium chloride, 10 HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), and 10 glucose (pH 7.4 adjusted with TEA-OH). The pipette solution contained (in mM) 95 CsOH, 95 aspartate, 40 CsCl, 4 MgCl2, 5 ethylene glycol tetraacetic acid, 2 disodium adenosine-5′-triphosphate, 5 HEPES, and 8 creatine phosphate (pH 7.2 adjusted with CsOH). To test the response induced by CnTX, the cells were treated with 100 nM CnTX for 3 min before current measuring.
Immunocytochemical staining
The cultured H295R cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. Then, the cells were washed in phosphate-buffered saline and incubated in 5% normal goat serum (Vector Labs, Burlingame, CA, USA) in phosphate-buffered saline at room temperature for 60 min to block the nonspecific sites. The cells were incubated with 1:100-diluted primary antibodies in phosphate-buffered saline (Cav2.2/α1B antibody, Alomone Labs, Jerusalem, Israel; Cav1.2/α1C antibody, Alomone Labs; and Cav3.1/α1G antibody, Sigma-Aldrich) overnight at 4 °C.27 After being rinsed, the cells were incubated with the secondary antibody (biotinylated anti-rabbit immunoglobulin G; Invitrogen) and the third antibody (streptavidin-conjugated Alexa Fluor 555; Invitrogen) for 1 h at room temperature. Negative controls for each procedure were obtained by replacing the primary antibody with purified nonimmune rabbit immunoglobulin G (1:1000). Images were captured using a phase-contrast microscope (Axiovert 100; Carl Zeiss, Oberkochen, Germany) with Pixera Viewfinder software (version 3.0; Pixera Corporation, Los Gatos, CA, USA).
Calcium imaging
H295R cells were seeded onto eight-well chamber slides and cultured until 80% confluence. Next, the medium was replaced with fresh medium containing 0.2% Nu-serum, and the cells were cultured for an additional 24 h. The cells were then loaded with 5 μM fura-2/AM (Invitrogen) for 40 min. This loading was performed in Hank's balanced salt solution containing 2.5 mM probenecid (Sigma-Aldrich), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Invitrogen), and 4% Pluronic F-127 (Invitrogen). The cells were rinsed and then incubated for 20 min in Hank's balanced salt solution with or without each calcium channel inhibitor. After the 20 min incubation, different concentrations of Ang II were added to the cells, and fluorescence images were recorded using a microscope (TMD-2S; Nikon, Tokyo, Japan) and a video-based imaging system (Aquacosmos; Hamamatsu Photonics, Shizuoka, Japan), which enabled controlled image acquisition and display. The ratio of the fluorescence intensity at 340 nm to the intensity at 380 nm was calculated to evaluate the change in intracellular calcium levels. To test the response induced by the activation of CnTX, the cells were stimulated with 100 nM Ang II solution at room temperature.
Small interfering RNA (siRNA) transfection and cell culture conditions
Transfection with siRNA of the human α1B subunit (Human CACNA1B, Thermo Fisher Scientific, Waltham, MA, USA) or of non-target siRNA (Thermo Fisher Scientific) was performed with DharmaFECT1 (Thermo Fisher Scientific). H295R cells were transfected in growth medium containing DharmaFECT1 reagent with 10 nM of siRNA for 16 h. Then, the cells were incubated for an additional 24 h in fresh medium containing 0.2% Nu-serum. The culture supernatants were replaced with medium containing 100 nM Ang II, and, after incubation for 48 h, the steroid content of the conditioned media and the protein content of the cells were determined.
Western blotting analysis of H295R cells
H295R cells were solubilized in RIPA buffer (pH 8.0: 1% NP-40, 1% SDS, 1% sodium deoxycholate, 25 mM Tris HCl. 150 mM NaCl) containing 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P40, 150 mM NaCl, 50 mM Tris-HCl, 1 mM phenylmethylsulfonyl fluoride, and 10 μg μl−1 leupeptin. Immunoprecipitated proteins were fractionated by 10% SDS-polyacrylamide gel electrophoresis and electrotransferred onto a polyvinylidene difluoride membrane. The blots were incubated with living colors polyclonal antibody (Alomone labs ACC-002) and visualized using West pico solution (Thermo Fisher Scientific).
Quantitative reverse transcription-PCR
Total RNA was isolated from H295R cells using the RNeasy kit (Qiagen ,Hilden, Germany) after 6,12, 24, and 48 h of incubation. Total RNA (1 μg) was reverse-transcribed using SuperScript III RT (Invitrogen). Quantitative real-time reverse transcription-PCR was performed using the 7500 Real-time PCR System with SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA, USA) and sequence-specific primers (Table 1). Complementary DNAs (Takara Bio, Shiga, Japan) from human tissues were also used as templates. The mRNA levels were normalized to the levels of glyceraldehyde-3-phosphate dehydrogenase and hypoxanthine–guanine phosphoribosyltransferase.
Statistical analyses
The results are expressed as the mean±s.e.m. The differences between the non-treated and control groups were evaluated using Student's t-test. The differences among the results for the Ang II-treated groups were evaluated using Dunnett's test or time-wise multiple comparisons of longitudinal data (SAS Institute, Cary, NC, USA).
Results
N-type calcium channels are expressed in H295R cells, and CnTX inhibits increases in Ang II-stimulated intracellular calcium level
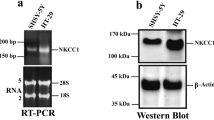
The reverse transcription-PCR data (Figure 1c) showed the gene expression of L-type, N-type, and T-type calcium channels in the H295R cells as well as in the brain and heart. In addition, the data from western blotting and immunocytochemical staining revealed the expression of N-type (Figures 1d and 2c, d), L-type (Figure 2e), and T-type calcium channels (Figure 2f) in the H295R cell line. The patch clamp recording revealed voltage-dependent barium currents inhibited by CnTX (100 nM; Figures 1a and b). As previously described, Ang II increased intracellular calcium levels in the H295R cells in a concentration-dependent manner (Figures 3a and b), and 100 nM CnTX inhibited 100 nM Ang II-induced the increased intracellular calcium level (75.4±4%, P<0.05; Figure 3c).

Expression of N-type calcium channels in H295R cells. (a) Inhibitory effect of 100 nM omega-conotoxin GVIA (CnTX) on voltage-dependent calcium channel (VDCC) currents in H295R cells. The representative traces for barium currents upon application of 30-ms test pulses from −50 to 50 mV with 10-mV increments starting from a Vh of −80 mV. (b) Current density–voltage (I–V) relationships of VDCCs. I–V relationships in control (non-treat), 100 nM CnTX-treated conditions (CnTX), and subtraction of barium currents in the presence of CnTX from control barium current (subtracted). Data were obtained from three independent experiments (n=9) and expressed as the mean±s.e.m. (c) mRNA expression of the N-type calcium channel in H295R cells and human tissue. The data represent calcium channel mRNA expression in human tissues and cells (mean). (d) Western blotting of N-type calcium channel in H295R cells. Total protein (600 μg) was loaded. The CaV2.2 immunoreactivity is identified by incubation with anti-CaV2.2 (1:200). Identical procedures except the anti-CaV2.2 subjected to peptide block (anti-CaV2.2+peptide) fail to detect specific immunoreactivity.

Immunocytochemical localization of the calcium channels in H295R cells. The cells were immunostained with Cav2.2/α1B antibody (c, d), Cav1.2/α1C antibody (e), Cav3.1/α1G antibody (f), or nonimmune immunoglobulin G (a, b). Positive staining for each antigen was detected by performing immunofluorescence analysis using Alexa Fluor 555 (a, c, × 40; b, d–f, × 20). The figure depicts the result of one representative experiment. Similar results were obtained in two additional experiments with different passages of cells.

Expression of N-type calcium channels in H295R cells and the functional role of these channels in angiotensin II (Ang II)-induced aldosterone production. (a) Intracellular Ca2+ level as a cellular response to Ang II stimulation (0.1–10 μM). (b) The peak intracellular Ca2+ level was dependent on Ang II concentration. The data represent the percentage values of the intensity of fluorescence in the stimulated cells, and these values were relative to the corresponding values in cells treated with 100 nM of Ang II. (c) Inhibitory effect of omega-conotoxin GVIA (CnTX) on Ang II-induced intracellular Ca2+ increment in H295R cells. The data represent the percentage values of the intensity of fluorescence in the incubated cells; these values were relative to the corresponding values for cells treated with 100 nM of Ang II. All the results are shown as the mean±s.e.m. (n=5–8). †††P<0.001 for non-treated H295R cells vs. control. *P<0.05 for the control group vs. each CnTX-treatment group.
N-type calcium channels have a role in corticosteroid production
Aldosterone and cortisol production in H295R cells stimulated with 100 nM Ang II for 48 h (aldosterone: 100±4%, Figure 4a; cortisol: 100±10%, Figure 4b) was four- to fivefold higher than in non-stimulated cells (aldosterone: 23±1%, P<0.001; cortisol: 17±3%, P<0.001). CnTX inhibited Ang II-induced aldosterone production (10 nM: 76±6%, P<0.01; 100 nM: 58±4%, P<0.001; 1000 nM: 52±5%, P<0.001; Figure 4a) and Ang II-induced cortisol production (10 nM: 65±9%, P<0.05; 100 nM: 61±11%, P<0.05; 1000 nM: 53±8%, P<0.01; Figure 4b) in a concentration-dependent manner. Next, we evaluated the effect of α1B RNA interference on Ang II-induced aldosterone production in H295R cells. In comparison with non-target siRNA, α1B siRNA caused a significant reduction in α1B mRNA expression during the entire investigation (0 h: 81±3%, P<0.01; 6 h: 52±1%, P<0.001; 12 h: 54±2%, P<0.001; 48 h: 42±7%, P<0.01; Figure 5a). The increase in aldosterone production was significantly inhibited by the siRNA for the N-type calcium channel (Figure 5b; 60±4%, P<0.05).

Inhibitory effect of N-type calcium channel blockers on corticosteroid production in H295R cells stimulated with 100 nM angiotensin II (Ang II). (a) Inhibitory effect of omega-conotoxin GVIA (CnTX) on aldosterone production in H295R cells stimulated with 100 nM Ang II. The bars represent the percentage values of the aldosterone secretion of culture supernatants (n=15). These values were normalized by the cellular protein content and were relative to the corresponding values for cells treated with 100 nM of Ang II (control; 19±2 pg/μg protein). (b) Inhibitory effect of CnTX on cortisol production in H295R cells stimulated with 100 nM Ang II. The bars represent the percentage values for cortisol secretion by the culture supernatant (n=8), and these values were normalized by the cellular protein content and were relative to the corresponding value for the cells treated with 100 nM Ang II (control; 2255±352 pg/μg protein). (c) Effect of 100 nM CnTX on the expression of CYP11B2 after stimulation with 100 nM Ang II (n=4). (d) Effect of 100 nM CnTX on the expression of CYP11B1 after stimulation with 100 nM Ang II (n=4). †††P<0.001 and †P<0.05 for the non-treated group vs. the control group. All results are shown as means±s.e.m. *P<0.05; **P<0.01; and ***P<0.001 for the control group vs. each CnTX-treatment group.

Effect of α1B knockdown on corticosteroid production and gene expression in H295R cells stimulated with 100 nM angiotensin II (Ang II). (a) In comparison with non-target small interfering RNA (siRNA), α1B siRNA caused a significant reduction in α1B mRNA expression during the entire investigation. These values were relative to the corresponding value for the cells treated with non-target siRNA (n=4). (b) The effect of α1B RNA interference on Ang II-induced aldosterone production in H295R cells. The increase in aldosterone production was significantly inhibited by the siRNA for the N-type calcium channel. The bars represent the percentage values normalized by the cellular protein content, and these values were relative to the corresponding value for the cells treated with non-target siRNA (n=5). (c) The effect of α1B RNA interference on the expression of CYP11B2 after stimulation with 100 nM Ang II (n=8). These values were relative to the corresponding value for the cells treated with non-target siRNA (n=8). (d) The effect of α1B RNA interference on the expression of CYP11B2 after stimulation with 100 nM Ang II (n=8). These values were relative to the corresponding value for the cells treated with non-target siRNA (n=8). †††P<0.001, and ††P<0.001 for the difference among the siRNA-non-treated groups. All results are shown as means±s.e.m. ###P<0.001, ##P<0.001, and ##P<0.001 for the difference among the siRNA-treated groups.
N-type calcium channels do not have a significant influence on the expression of steroidogenic enzymes
We assessed the expression of the mRNAs encoding the steroidogenic enzymes CYP11B2 (aldosterone synthase) and CYP11B1 (11β-hydroxylase, the enzyme catalyzing the last step in cortisol synthesis). Ang II (100 nM) caused transient and acute induction of CYP11B2 (Figure 4c) and CYP11B1 (Figure 4d) mRNA expression. The CYP11B2 and CYP11B1 mRNA levels peaked after 12 h (215±6%, P<0.001) and 6 h (146±8%, P<0.001) of Ang II stimulation, respectively. These results were consistent with the findings of a previous study.28 CnTX at 100 nM failed to suppress the Ang II-induced increase in the levels of CYP11B2 and CYP11B1 transcripts but inhibited aldosterone and cortisol production (Figures 4c and d). In addition, α1B interference had no effect on the Ang II-induced increase in CYP11B1 levels after 6 h of Ang II stimulation (Figure 5d) but increased CYP11B2 mRNA expression in comparison with non-target siRNA treatment after 12 h of Ang II stimulation (123±4%, P<0.001: Figure 5c).
Each CCB has a different effect on corticosteroid production, and this action is dependent on the type of blocked calcium channel
Each CCB inhibited Ang II-induced aldosterone production in a concentration-dependent manner (Figure 6a). Cilnidipine and efonidipine significantly suppressed Ang II-induced aldosterone production at a low concentration (⩽10 nM), as was observed in the case of CnTX. Moreover, they were significantly more potent than nifedipine (at 10 nM: cilnidipine, 65±4%, P<0.001; efonidipine, 72±6%, P<0.01; nifedipine, 85±5%, P>0.05). Similarly, each CCB inhibited Ang II-induced cortisol production in a concentration-dependent manner (Figure 6b). The cilnidipine and efonidipine concentrations required to inhibit Ang II-induced production of cortisol were lower than the nifedipine concentration required to achieve the same effect (at 100 nM: cilnidipine, 68±9%, P<0.05; efonidipine, 68±6%, P<0.05; nifedipine, 65±9%, P>0.05).

Inhibitory effect of each CCB on corticosteroid production and gene expression in H295R cells stimulated with 100 nM Ang II. (a) Each CCB has a different action on aldosterone secretion. The bars represent the percentage values normalized by the cellular protein content (n=12). These values were relative to the corresponding value for the cells treated with 100 nM angiotensin II (Ang II; control; 18±3 pg/μg protein). (b) Each CCB has a different action on cortisol secretion, and this action depends on the type of the blocked calcium channel. The bars represent the percentage values normalized by the cellular protein content (n=8), and these values were relative to the corresponding value for the cells treated with 100 nM Ang II (control; 2255±352 pg/μg protein). (c) The effect of each CCB (100 nM) on the expression of CYP11B2 after stimulation with 100 nM Ang II (n=4). The bars represent the percentages of CYP11B2 mRNA expression, which were normalized and were relative to the corresponding value for the control (Ang II, 100 nM). (d) The effect of each CCB (100 nM) on the expression of CYP11B1 after stimulation with 100 nM Ang II (n=4). The bars represent the percentages of CYP11B1 mRNA expression, which were normalized and were relative to the corresponding value for the control (Ang II, 100 nM). †††P<0.001 for the non-treated group vs. the control group. *P<0.05; **P<0.01; and ***P<0.001 for the control group vs. each CCB group. CCB, calcium channel blocker; Cil, cilnidipine; Efo, efonidipine; Nif, nifedipine.
Effects of CCBs on the gene expression of steroidogenic enzymes
Next, we examined the impact of the blockade of different types of calcium channels (100 nM) on Ang II-induced CYP11B2 and CYP11B1 mRNA expression. After 12 h of Ang II stimulation, the mRNA expression of CYP11B2 was suppressed by all of the CCBs (cilnidipine, 83±0%; nifedipine, 79±2%; efonidipine, 71±3%: P<0.001; Figure 6c). In contrast, after 6 h of Ang II stimulation, the expression of CYP11B1 mRNA was suppressed by nifedipine (75±5%, P<0.01; Figure 6d) and efonidipine (67±1%, P<0.001; Figure 6) but not by cilnidipine (88±5%; Figure 6d). However, all of the calcium channel blockers inhibited Ang II-induced cortisol production (Figure 6b).
Discussion
This study is the first report on the distribution of N-type calcium channels in human adrenocortical cells and the involvement of these channels in the Ang II-induced calcium influx in these cells. Furthermore, the data from experiments with CnTX and N-type calcium channel siRNA suggest that N-type calcium channels are involved in Ang II-induced aldosterone production. Previous studies showed that Ang II induces aldosterone production via the generation of cytoplasmic calcium signaling,7 and the Ang II effects were mimicked by calcium ionophore A23187 and blocked by CCBs,29, 30 suggesting the importance of calcium influx inhibition in the suppression of Ang II-induced aldosterone production. These reports suggest that the suppressive effect of N-type calcium channel blockers on Ang II-induced corticosteroid production may be related to their inhibitory activity against the calcium influx induced by Ang II, as in the case of T-type calcium channel blockers. However, the blockade of N-type calcium channels failed to suppress the expression of CYP11B2 mRNA induced by Ang II, although the aldosterone-production mechanism mediated by T-type and L-type calcium channels is accompanied by an increase in CYP11B2 mRNA levels, as previously reported.31 The reason for the differences in the expression level of the CYP11B2 gene is still unclear because the number of factors influencing aldosterone secretion was greater than the number of factors influencing CYP11B2 mRNA expression. For example, aldosterone production is regulated by both CYP11B2 expression and CYP11B2 enzyme activity.30, 32 Lisurek and Bernhardt32 suggested that CYP11B2 enzyme activity may be post-translationally controlled by protein phosphorylation, which exerts short-term control over CYP11B2 activity. One mechanism that can be hypothesized on the basis of these reports suggests that N-type calcium channels regulate aldosterone production by changing the activity of steroidogenic enzymes.
We also investigated whether the effects of CCBs on Ang II-induced corticosteroid production and on the expression of steroidogenic-enzyme genes in H295R cells depend on the type of blocked channels. To date, in vivo33, 34 and in vitro29, 35 studies have revealed differences among the corticosteroid production-inhibiting effects of CCB. In fact, our study indicated that each CCB succeeded in suppressing Ang II-induced corticosteroid production, and low concentrations of cilnidipine and efonidipine inhibited Ang II-induced production of corticosteroids. Nifedipine was much less potent in inhibiting Ang II-induced aldosterone production (⩾100 nM), although it suppressed Ang II-induced mRNA expression of CYP11B2 with the same potency as the other CCBs. These results suggest that the suppression of Ang II-induced CYP11B2 mRNA expression by L-type CCBs may partially contribute to the inhibition of aldosterone production. The N-type or T-type calcium channel blockade may have primary roles in the inhibition of Ang II-induced aldosterone production. In addition, cilnidipine failed to suppress CYP11B1 mRNA expression induced by 100 nM Ang II, although cilnidipine and efonidipine significantly inhibited cortisol production induced by 100 nM Ang II. These results suggest that L-type and T-type calcium channels control corticosteroid production by regulating CYP11B1 and CYP11B2 mRNA expression, and N-type calcium channels may offer another way of controlling corticosteroid production in adrenocortical cells. Also, cilnidipine may have an effect on CYP11B1 expression because of its blockade action of L-type calcium channel. Our results contradict earlier results that suggested that cilnidipine and nifedipine may not suppress aldosterone production in H295R cells.29 However, some differences exist in the methods used in the previous study and in our study. In particular, the most important aspect is the difference in the evaluation time between the earlier study (24 h) and our study (48 h). We assessed changes in aldosterone production over time to determine an appropriate evaluation time. The result suggested that 24 h may be too short to recognize the effect of the N-type calcium channel blockade on aldosterone production (data not shown); therefore, the variation in data between the previous report and the current results may reflect a difference of evaluation period.
In this study, the cilnidipine concentrations required to suppress the Ang II-induced aldosterone production were less than, or within the range of, therapeutic plasma concentrations (0.8–32.5 nM),36 which suggests that cilnidipine may have potential therapeutic applications in patients with chronic heart failure and chronic kidney disease.
Conclusions
N-type calcium channels are expressed in H295R human adrenocortical cells, and Ang II-induced aldosterone production is mediated by T-type and L-type calcium channels as well as by N-type calcium channels. Moreover, our data indicate that the N-type calcium channels contribute minimally to the Ang II-induced expression of CYP11B1 and CYP11B2 mRNAs. These findings suggest that N-type calcium channels may have a role in aldosterone production in adrenocortical cells.
References
Goodfriend TL . Aldosterone—a hormone of cardiovascular adaptation and maladaptation. J Clin Hypertens (Greenwich) 2006; 8: 133–139.
Brown NJ . Aldosterone and end-organ damage. Curr Opin Nephrol Hypertens 2005; 14: 235–241.
Bianchi S, Bigazzi R, Campese VM . Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70: 2116–2123.
Mizuno Y, Yoshimura M, Yasue H, Sakamoto T, Ogawa H, Kugiyama K, Harada E, Nakayama M, Nakamura S, Ito T, Shimasaki Y, Saito Y, Nakao K . Aldosterone production is activated in failing ventricle in humans. Circulation 2001; 103: 72–77.
Yamamoto N, Yasue H, Mizuno Y, Yoshimura M, Fujii H, Nakayama M, Harada E, Nakamura S, Ito T, Ogawa H . Aldosterone is produced from the ventricles of patients with essential hypertension. Hypertension 2002; 39: 958–962.
Mizuno Y, Yasue H, Yoshimura M, Harada E, Fujii H, Nakamura S, Yamamoto N, Ogawa H, Nakao K . Adrenocorticotropic hormone is produced in the ventricle of patients with essential hypertension. J Hypertens 2005; 23: 411–416.
Spät A, Hunyady L . Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev 2004; 84: 489–539.
Natke Jr E, Kabela E . Electrical responses in cat adrenal cortex: possible relation to aldosterone secretion. Am J Physiol 1979; 237: E158–E162.
Aguilera G, Catt KJ . Participation of voltage-dependent calcium channels in the regulation of adrenal glomerulosa function by angiotensin II and potassium. Endocrinology 1986; 118: 112–118.
Rossier MF, Burnay MM, Vallotton MB, Capponi AM . Distinct functions of T- and L-type calcium channels during activation of bovine adrenal glomerulosa cells. Endocrinology 1996; 137: 4817–4826.
Takahara A . Cilnidipine: a new generation ca channel blocker with inhibitory action on sympathetic neurotransmitter release. Cardiovasc Ther 2009; 27: 124–139.
Morad M, Davies NW, Kaplan JH, Lux HD . Inactivation and block of calcium channels by photo-released Ca2+ in dorsal root ganglion neurons. Science 1988; 241: 842–844.
Patil PG, Brody DL, Yue DT . Preferential closed-state inactivation of neuronal calcium channels. Neuron 1998; 20: 1027–1038.
Dubel SJ, Starr TV, Hell J, Ahlijanian MK, Enyeart JJ, Catterall WA, Snutch TP . Molecular cloning of the α-1 subunit of an ω-conotoxin-sensitive calcium channel. Proc Natl Acad Sci 1992; 89: 5058–5062.
Jeong SW, Wurster RD . Calcium channel currents in acutely dissociated intracardiac neurons from adult rats. J Neurophysiol 1997; 77: 1769–1778.
Smith PA . N-type Ca2+-channels in murine pancreatic {beta}-cells are inhibited by an exclusive coupling with somatostatin receptor subtype 1. Endocrinology 2009; 150: 741–748.
Wykes RC, Bauer CS, Khan SU, Weiss JL, Seward EP . Differential regulation of endogenous N- and P/Q-type Ca2+ channel inactivation by Ca2+/calmodulin impacts on their ability to support exocytosis in chromaffin cells. J Neurosci 2007; 27: 5236–5248.
Dunlap K, Luebke JI, Turner TJ . Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci 1995; 18: 89–98.
Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D . Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 2004; 41: 127–138.
Khanna R, Zougman A, Stanley EF . A proteomic screen for presynaptic terminal N-type calcium channel (CaV2.2) binding partners. J Biochem Mol Biol 2007; 40: 302–314.
Konda T, Enomoto A, Aritomi S, Niinuma K, Koganei H, Ogawa T, Nitta K . Different effects of L/N-Type and L-Type calcium channel blockers on the renin-angiotensin-aldosterone system in SHR/Izm. Am J Nephrol 2009; 30: 155–161.
Takahara A, Nakamura Y, Wagatsuma H, Aritomi S, Nakayama A, Satoh Y, Akie Y, Sugiyama A . Long-term blockade of L/N-type Ca(2+) channels by cilnidipine ameliorates repolarization abnormality of the canine hypertrophied heart. Br J Pharmacol 2009; 158: 1366–1374.
Rainey WE, Saner K, Schimmer BP . Adrenocortical cell lines. Mol Cell Endocrinol 2004; 228: 23–38.
Suzuki J, Otsuka F, Inagaki K, Takeda M, Ogura T, Makino H . Novel action of activin and bone morphogenetic protein in regulating aldosterone production by human adrenocortical cells. Endocrinology 2004; 145: 639–649.
Siriannj R, Carr BR, Pezzi V, Rainey WE . A role for src tyrosine kinase in regulating adrenal aldosterone production. J Mol Endocrinol 2001; 26: 207–215.
Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K . Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem 1998; 273: 34857–34867.
Liu J, Li XD, Ora A, Heikkijä P, Vaheri A, Voutilainen R . cAMP-dependent protein kinase activation inhibits proliferation and enhances apoptotic effect of tumor necrosis factor-alpha in NCI-H295R adrenocortical cells. J Mol Endocrinol 2004; 33: 511–522.
Romero DG, Plonczynski M, Vergara GR, Gomez-Sanchez EP, Gomez-Sanchez CE . Angiotensin II early regulated genes in H295R human adrenocortical cells. Physiol Genomics 2004; 19: 106–116.
Akizuki O, Inayoshi A, Kitayama T, Yao K, Shirakura S, Sasaki K, Kusaka H, Matsubara M . Blockade of T-type voltage-dependent Ca2+ channels by benidipine, a dihydropyridine calcium channel blocker, inhibits aldosterone production in human adrenocortical cell line NCI-H295R. Eur J Pharmacol 2008; 584: 424–434.
Romero DG, Plonczynski MW, Gomez-Sanchez EP, Yanes LL, Gomez-Sanchez CE . RGS2 is regulated by angiotensin II and functions as a negative feedback of aldosterone production in H295R human adrenocortical cells. Endocrinology 2006; 147: 3889–3897.
Rossier MF, Python CP, Capponi AM, Schlegel W, Kwan CY, Vallotton MB . Blocking T-type calcium channels with tetrandrine inhibits steroidogenesis in bovine adrenal glomerulosa cells. Endocrinology 1993; 132: 1035–1043.
Lisurek M, Bernhardt R . Modulation of aldosterone and cortisol synthesis on the molecular level. Mol Cell Endocrinol 2004; 215: 149–159.
Tanaka T, Tsutamoto T, Sakai H, Fujii M, Yamamoto T, Horie M . Comparison of the effects of efonidipine and amlodipine on aldosterone in patients with hypertension. Hypertens Res 2007; 30: 691–697.
Ishimitsu T, Kameda T, Akashiba A, Takahashi T, Ohta S, Yoshii M, Minami J, Ono H, Numabe A, Matsuoka H . Efonidipine reduces proteinuria and plasma aldosterone in patients with chronic glomerulonephritis. Hypertens Res 2007; 30: 621–626.
Bird IM, Mathis JM, Mason JI, Rainey WE . Ca(2+)-regulated expression of steroid hydroxylases in H295R human adrenocortical cells. Endocrinology 1995; 136: 5677–5684.
Kuji N, Minamisawa K, Yamakado M, Tochikubo O, Ishii M . Antihypertensive effect and pharmacokinetics of FRC-8653 (Cilnidipine) on hypertensive patients with renal impairment or normal renal function. Jpn Pharmacol Ther 1993; 21 (Suppl 1): 193–205 (in Japanese).
Acknowledgements
This study was supported by Ajinomoto Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Aritomi, S., Wagatsuma, H., Numata, T. et al. Expression of N-type calcium channels in human adrenocortical cells and their contribution to corticosteroid synthesis. Hypertens Res 34, 193–201 (2011). https://doi.org/10.1038/hr.2010.191
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2010.191
Keywords
This article is cited by
-
Cardiac hypertrophy in chronic kidney disease—role of Aldosterone and FGF23
Renal Replacement Therapy (2018)
-
L/N-type Ca2+ channels blocker cilnidipine ameliorated the repolarization abnormality in a chronic hemodialysis patient
Heart and Vessels (2017)
-
Ablation of the N-type calcium channel ameliorates diabetic nephropathy with improved glycemic control and reduced blood pressure
Scientific Reports (2016)
-
Long-term effects of L- and N-type calcium channel blocker on uric acid levels and left atrial volume in hypertensive patients
Heart and Vessels (2016)
-
L/N-type calcium channel blocker cilnidipine reduces plasma aldosterone, albuminuria, and urinary liver-type fatty acid binding protein in patients with chronic kidney disease
Heart and Vessels (2013)
