
Abstract
With methods for sequencing thousands of loci for many individuals, phylogeographic studies have increased inferential power and the potential for applications to new questions. In songbirds, strong patterns of inter-chromosomal synteny, the published genome of a songbird and the ability to obtain thousands of genetic loci for many individuals permit the investigation of differentiation between and diversity within lineages across chromosomes. Here, we investigate patterns of differentiation and diversity in Certhia americana, a widespread North American songbird, using next-generation sequencing. Additionally, we reassess previous phylogeographic studies within the group. Based on ~30 million sequencing reads and more than 16 000 single-nucleotide polymorphisms in 41 individuals, we identified a strong positive relationship between genetic differentiation and chromosome size, with a negative relationship between genetic diversity and chromosome size. A combination of selection and drift may explain these patterns, although we found no evidence for selection. Because the observed genomic patterns are very similar between widespread, allopatric clades, it is unlikely that selective pressures would be so similar across such different ecological conditions. Alternatively, the accumulation of fixed differences between lineages and loss of genetic variation within lineages due to genetic drift alone may explain the observed patterns. Due to relatively higher recombination rates on smaller chromosomes, larger chromosomes would, on average, accumulate fixed differences between lineages and lose genetic variation within lineages faster, leading to the patterns observed here in C. americana.
Similar content being viewed by others


Introduction
Multilocus investigations of genetic structure have recently become commonplace in phylogeographic studies. With new methods for obtaining reduced-representation libraries of the genome (for example, restriction digest-based methods, Miller et al., 2007 and ultraconserved elements, Faircloth et al., 2012) for many individuals, phylogeographic studies may contain thousands of loci with dozens of individuals sampled. In songbirds, strong patterns of interchromosomal synteny (Kawakami et al., 2014), the published genome of the Zebra Finch (Taeniopygia guttata; Warren et al., 2010) and thousands of genetic markers across the genome allow the opportunity to investigate not only phylogeographic structure in a clade well known for its high levels of geographic differentiation (Manthey et al., 2011a), but also diversity and differentiation across chromosomes (Manthey and Spellman, 2014).
Studies of the Chicken (Gallus gallus) and Zebra Finch genomes have identified biased recombination toward the telomeres of chromosomes (ICGSC, 2004; Backström et al., 2010); because bird chromosomes vary greatly in size (from kilobases to hundreds of megabases), higher than expected recombination near the telomeres causes smaller chromosomes to have a higher mean recombination rate than larger chromosomes. Additionally, recombination rates will generally scale negatively with chromosome length due to meiotic crossover requirements (Lynch, 2007). A recent study of the Chicken genome by Mugal and Nabholz (2013) identified a positive relationship between local recombination rate and genetic diversity, and a negative relationship between lineage divergence and recombination rate, similar to patterns found in humans (Keinan and Reich, 2010). Also, studies have identified patterns of diversity and divergence linked with the interactions of recombination rates and either directional selection (for example, Begun and Aquadro, 1992, Aguadé and Langley, 1994) or background selection (Nachman, 2001), illuminating a link between recombination rates and diversity and divergence. These findings suggest that, in general, larger chromosomes will tend to exhibit lower genetic diversity within and higher genetic divergence between lineages compared with smaller chromosomes. In White-throated Sparrows (Zonotrichia albicollis), a large multilocus study (~37 markers) supported this idea, with larger chromosomes exhibiting reduced genetic diversity (Huynh et al., 2010). In contrast, genomic studies of two Ficedula species identified no clear patterns of differentiation or diversity between chromosomes of different sizes (Ellegren et al., 2012). Rather, they identified genomic islands of divergence, in which small regions of each chromosome showed large spikes in FST between lineages and decreased diversity within lineages, presumably because of selection against introgression between species at these loci.
More broadly, birds tend to exhibit increased differentiation and reduced genetic diversity on the Z chromosome relative to autosomes (Carling and Brumfield, 2008; Balakrishnan and Edwards, 2009; Ellegren et al., 2012; Rheindt et al., 2013). This may be expected if there are genetic regions under selection on the Z chromosome (for example, to reduce expression of recessive alleles in hemizygous individuals); however, if lineages are diverging largely by genetic drift, the Z chromosome may simply act like a macrochromosome (albeit with a smaller effective population size). In this situation, we might expect the Z chromosome to show reduced diversity and increased differentiation between lineages relative to all autosomes pooled, but not other macrochromosomes in general (after correcting for NE).
The Brown Creeper (Certhia americana) is a widespread songbird of North America and represents a good study system to investigate chromosomal patterns of differentiation. Because it is a songbird, sequence data may be BLASTed to the Zebra Finch genome to identify upon which chromosome loci are found. There are two major lineages, split at 32°N latitude, within C. americana identified with both mitochondrial (mtDNA; Manthey et al., 2011a) and nuclear DNA (nDNA; Manthey et al., 2011b). Between the major lineages, there is also apparent quicker differentiation and reduced gene flow on the Z chromosome relative to autosomal loci (Manthey and Spellman, 2014). Finally, there is discordance between mtDNA and nDNA relationships between clades in the northern lineage (Manthey et al., 2011a, b); with thousands of loci across the genome, this discordance may be disentangled.
Here, using large panels of single-nucleotide polymorphisms (SNPs) across the genome, we investigate chromosomal patterns of divergence between and diversity within major lineages of C. americana, as well as reassess phylogeographic patterns across all major clades of the species. Using these data, we examine the following questions: (1) Is chromosomal genetic differentiation between lineages related to chromosome size? (2) Is chromosomal genetic diversity within lineages related to chromosome size? (3) Does the Z chromosome exhibit higher differentiation between lineages than would be expected based on chromosome size? (4) What are the phylogeographic relationships between populations based on thousands of loci? (5) Can accounting for genomic patterns in a phylogeographic context help inform population genomic processes?
Materials and methods
Sampling, laboratory procedures and SNP data set creation
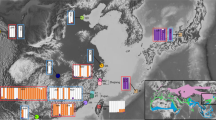
Tissue samples of 41 C. americana individuals were obtained from eight localities (Figure 1, Table 1, Supplementary Table 1), representing the structured clades recovered in previous mtDNA analyses (Manthey et al., 2011a). Total genomic DNA was extracted from tissue samples using a QIAGEN (Hilden, Germany) DNeasy tissue extraction kit following the manufacturer protocols. DNA quality and quantity was determined using agarose gel electrophoresis and a NanoDrop Spectrophotometer (ND-1000, Thermo Fisher, Inc., Lenexa, KS, USA). Restriction-site associated DNA sequencing of the samples followed the protocols described by Parchman et al. (2012), with a detailed protocol available from Dryad-10.5061/dryad.m2271pf1. Because the protocol is described in detail in Parchman et al., we provide only the major steps involved in the protocol.

Sampling map (a) and phylogeographic relationships (b) inferred from the SNP data set inclusive of a minimum of 30% of individuals for each locus (i.e., 30% coverage data matrix). Sampling letters match descriptions in Table 1. All asterisks at nodes in (b) indicate support >0.95 in SNAPP phylogenetic analyses for all SNP data sets. The asterisk with an arrow indicates a node supported strongly only by the 30% SNP data set (other data sets posterior probability=0.85). All STRUCTURE results identified hierarchical genetic structure for each data set, separating northern and southern populations with 100% assignment to either cluster. Secondary-level STRUCTURE results are shown below phylogeny (north k=2, south k=2 or 3), with each bar representing an individual.
Samples were first subjected to restriction digestion, using the restriction enzymes EcoRI and MseI, followed by adaptor ligation. Adaptor sequences included the Illumina sequencing adaptor, an 8- to 10-bp individual bar code on the EcoRI side of the fragment, followed by additional bases that match the restriction cut site sequence. Individual bar code sequences used for this study differed by at least four base pairs and can be found in Supplementary Table S1. Restriction reactions were accomplished in 9 μl reactions and incubated at 37 °C for 8 h. Ligation reactions were performed in 11 μl reactions, incubated at 16 °C for 6 h and quenched with 189 μl of 0.1 × TE buffer. Each sample was then PCR amplified using the Illumina PCR primers (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′ and 5′-CAAGCAGAAGACGGCATACGAGCTCTTCCGATCT-3′) that were complementary to the ligated adaptor sequences. Two 20 μl reactions were set up for each sample. The PCR master mix for each reaction contained the following: 9.6 μl UltraPure water, 4 μl 5 × Iproof Buffer, 0.4 μl 10 mM dNTPs, 0.4 μl 50 mM MgCl2, 1.33 μl 5 μM Illumina Primers, 0.2 μl DMSO, 0.15 μl Iproof (Bio-Rad, Inc., Hercules, CA, USA) TAQ polymerase. Amplification conditions consisted of the following: 98 °C for 30 s; 30 cycles of: 98 °C for 20 s, 60 °C for 30 s, 72 °C for 40 s, and final extension at 72 °C for 10 min. PCR products from all samples were pooled into a single sample. Before agarose gel electrophoresis was performed pooled PCR product was concentrated using vacuum centrifugation (200 μl pooled PCR product was transferred to each of 5 microcentrifuge tubes, placed into the Eppendorf Vacufuge and spun until only 100 μl of product remained, ~40 min). A volume of 50 μl of the pooled and concentrated PCR product was placed into each of 12 lanes of a 2% agarose gel and subjected to electrophoresis at 70 V for 1.5 h. The 300- to 400-bp region of the gel was excised and purified using a QIAquick gel purification kit (QIAGEN Inc.). Quality and quantity of the DNA fragment library was estimated using a NanoDrop Spectrophotometer (Thermo Fisher, Inc.) and a Bioanalyzer (Agilent, Inc., Santa Clara, CA, USA). DNA was sequenced on an Illumina (San Diego, CA, USA) HiSeqTM2000 using a single end 1 × 100 module at the University of Wisconsin Biotechnology Center DNA Sequencing Facility.
The STACKS (Catchen et al., 2013) pipeline was used to assemble loci de novo from the data obtained from the Illumina sequencing run. The included process_RADtags python script was used for quality control and assignment of sequencing reads to individuals. We set the quality threshold (for sequences to be included downstream) as greater than an average phred score of 10 in sliding windows of 15 base pairs and removed sequences with possible adapter contamination, while allowing for barcode rescue. Within STACKS, the ustacks, cstacks and sstacks commands were used to create libraries of loci: one for each individual and one for all loci shared among individuals. We used ustacks and sstacks with the default settings, while modifying the allowed number of mismatches (five) between samples because of possible deep divergence in some loci between C. americana individuals from different lineages.
Also within STACKS, the populations module was used to create three data sets that consisted of 30, 40 and 50% locus × individual matrices from the northern and southern lineages (that is, 30, 40 or 50% coverage data set; Figure 1). Although estimated absolute values of population genetic parameters may not be accurate due to incomplete matrices, the strong correlation between true and estimated values in simulations (Arnold et al., 2013) suggests we will be able to obtain general trends even from a 30% coverage matrix (r~0.8 in simulations). To preclude use of potentially paralogous loci, those with greater than 10 SNPs or with observed heterozygosity greater than 50% were removed from each data set using custom R (R Development Core Team, 2012) scripts. Following removal of loci, the populations module of STACKS was rerun to estimate pairwise FST values and within population nucleotide diversity (π) for each data set (see Catchen et al., 2013 for population genetic statistics formulae).
To investigate whether selection might be acting on particular genetic loci, we conducted an outlier analysis in BayeScan (Foll and Gaggiotti, 2008). BayeScan compares the posterior probabilities of two models, one using a neutral model with a population-level FST shared across all loci and another that incorporates locus-specific FST estimates (the selection component) to explain observed differences in allele frequencies. If the model invoking selection is necessary to describe variation in allele frequencies, then that locus is assumed to depart from neutrality. Following 20 pilot runs, BayeScan was run for a burn-in period of 50 000 iterations, followed by an additional 50 000 iterations sampled every 10; the FIS distribution and prior odds for the neutral model were used with default settings. BayeScan was performed on the data set with all individuals, as well as data sets containing only individuals in the northern or southern lineages.
Investigation of phylogeographic structure
We used the program STRUCTURE (Pritchard et al., 2000) to investigate levels of population structure without a priori input using the three SNP data sets. For each data set, we inferred lambda by estimating the likelihood of one population (k=1) and allowing lambda to converge. Subsequent runs of STRUCTURE used a fixed lambda inferred from the initial run and the admixture model. We ran five replicates of STRUCTURE for values of k=1–8, using a burn-in period of 50 000 steps followed by 150 000 MCMC iterations. We used the ΔK method of Evanno et al. (2005) to estimate the true number of populations from STRUCTURE output. While the method of Evanno et al. (2005) cannot be calculated at k=1, the likelihood of the data at k=1 excluded it as a possibility (Supplementary Information 1). For all data sets, the STRUCTURE analysis found k=2 (splitting all northern from southern populations with 100% assignment; see also Results); we therefore split the data sets into two parts and repeated the same steps as above for the separate data sets with values of k=1–5. This was done to infer whether there was weaker, more fine-scale genetic structure within each of the data sets.
We estimated a species tree using the likelihood method of Bryant et al. (2012) implemented in SNAPP, an add-on to BEAST v.2 (Bouckaert et al., 2014). Because this method requires unlinked SNPs, we pruned our data sets to include only the first SNP of each locus. For all data sets, we used default priors for theta as implemented in SNAPP, while inputting empirical estimates of mutation rates identified from major and minor allele base frequencies in the data set. Two independent SNAPP runs were performed for each data set. Stationarity of each SNAPP analysis was evaluated using two approaches (Supplementary Information 2): (1) we used TRACER (Rambaut and Drummond, 2007) to identify plateauing of posterior and likelihood estimates; and (2) we examined the cumulative posterior probabilities of tree bipartitions using the ‘cumulative’ utility implemented in AWTY (Nylander et al., 2008). The burn-in was defined as 200 000 MCMC generations, as determined by stationarity of all data sets before that point. Following burn-ins, each independent run was continued for an MCMC chain length of 1.4 million generations, with parameters and trees sampled every 1000. We assessed convergence of independent runs for each data set by comparing bipartition posterior probabilities between independent analyses (Supplementary Information 2). Replicate tree files for each data set were combined, summarized and visualized using Logcombiner v1.7.5, Tree Annotator v1.7.5 and FigTree v1.4, respectively, all of which are included in the BEAST v.2 (Bouckaert et al., 2014) package. Finally, we used custom R (R Development Core Team, 2012) scripts to identify fixed differences between the north and south lineages.
Chromosomal levels of variation
The BLAST+ utility (Camacho et al., 2009) was used to match loci in our study with chromosomal locations in the Zebra Finch (Taeniopygia guttata) genome, which is possible because of strong patterns of synteny in birds (Kawakami et al., 2014). Although we are able to match loci to chromosomes, there have been many intrachromosomal recombination events (for example, inversions; Kawakami et al. 2014) precluding locating the position of a locus on a chromosome. Although this is true, we mapped all loci and fixed differences to Zebra Finch chromosomal location to explore potential clustering of genetic variation (Supplementary Information 4). Loci were considered a match to the Zebra Finch genome if they had 70% sequence identity, and a maximum e-value of 0.01. To test robustness of this e-value, we BLASTed samples with maximum e-values of 0.001 and 0.0001. The number of loci matching each chromosome was strongly related between data sets (R2>0.999), so we proceeded with the e-value<0.01 results. For each chromosome (30% coverage data set), all matched loci were rerun in STACKS to obtain FST estimates between and nucleotide diversity estimates within the northern and southern C. americana lineages. To examine relationships between chromosome size and population genetic estimates (FST and π), we performed linear regression using R.
Results
Properties of sequence data and SNP data sets
From 41 individuals on a partial Illumina HiSeq2000 lane, we obtained 30 766 853 sequence reads. The number of reads per individual was highly variable, ranging from 188 613 to 1 115 491 (mean=750 411; median=765 466; standard deviation=225 772). Following trimming of individual barcodes and the restriction site, sequencing resulted in a total of ~2.6 billion base pairs. There were 16 044 SNPs in our 30% coverage data set, with a mean of ~1.2 SNPs per locus (Table 2; Supplementary Table 2). The majority of SNPs in all coverage data sets were variable only within one of the two main lineages (Figure 2). Coverage was consistent across chromosomes (Figure 3a, Supplementary Table 3), with the Z-chromosome underrepresented. This underrepresentation was not likely due to polymorphism between lineages in the restriction sites; when the populations module of STACKS was rerun for each lineage separately, the Z-chromosome was still underrepresented in both lineages (Supplementary Table 4).

Sources of variability for the 30, 40 and 50% coverage data sets. The majority of the variation is due to polymorphisms only variable within one of the lineages (private north or south). Less than 25% of each data set’s SNPs were shared polymorphisms or fixed differences.

Number of loci (a) and number of loci with fixed differences (b) plotted against chromosome size. P-values for both plots are <0.001. Arrows point to the Z-chromosome. Data are from the 30% coverage data set (i.e., 30% of loci × individuals data matrix).
To determine significance of selection tests performed in BayeScan, we interpreted the LPOR (log posterior odds ratio) using Jeffreys’ scale of evidence (Jeffreys, 1961), with a value of one as strong evidence of selection. The maximum observed value among loci was −0.362, which was not close to significant for any loci. This pattern was also observed in data sets limited to the northern (max LPOR=−0.107) or southern lineage (max LPOR=0.441). A possible explanation for lack of evidence for selection may be due to the high background FST level, as the authors of BayeScan (Foll and Gaggiotti, 2008) suggest that high average genetic differentiation lessens the power of BayeScan to detect outlier loci.
Phylogeographic structure
All population pairwise FST values are presented in Supplementary Table 5 and summarized in Figure 4 as pairwise comparison means within a lineage or between lineages. All FST pairwise comparisons between lineages identified high levels of differentiation between northern and southern populations (Figure 4; Supplementary Table 5). Within lineages, FST pairwise comparisons were generally higher in the southern lineage (Figure 4; Supplementary Table 5), while the eastern population had the highest pairwise FST comparisons within the northern lineage. In the 30% coverage data set, ~10% of SNPs were fixed between lineages, though the proportion lessened with more comprehensive SNP coverage (Figure 2; Supplementary Table 6). Based on BLAST+ results, fixed differences were spread evenly across the genome (Figure 3b; Supplementary Table 3).

Summary of all pairwise FST comparisons. Each dot and confidence interval represent the mean and standard deviation of all comparisons within or between identified lineages as in Figure 1b (e.g., East (A) pairwise FST values within (northern pairwise comparisons) and between (southern pairwise comparisons) lineages). Northern populations are labeled (A–D) and southern populations are labeled (E–H), as shown in the sampling map in Figure 1.
For each of the SNP coverage data sets, we recovered hierarchical genetic structure from STRUCTURE analyses (Figure 1b; Supplementary Information 1), with strong support for 100% assignment to the northern or southern lineage (that is, K=2). Within the southern lineage, STRUCTURE analyses identified either two or three population groups (Figure 1b; Supplementary Information 1); these results support strong structuring of the Chiapas and Nuevo Leon populations, with the Morelos and Jalisco populations either being intermediate between the two (at k=2) or with strong support as an independent population unit (at k=3). Within the northern lineage, we recovered two populations, with strong support for an east-west split (Figure 1b; Supplementary Information 1).
Independent SNAPP analyses for all variable-coverage SNP data sets recovered the same topology, with only minor differences in support values (Figure 1b). The basal split supports the previously identified north-south separation. Within the northern lineage, the eastern localities are separated from western localities, with no support for relationships between western localities. In the southern lineage, individuals from Chiapas are split from all other Mexican birds. Consistent, but weaker, support is identified for the split between the Nuevo Leon population from the Morelos and Jalisco birds.
Chromosomal patterns
We identified a strong relationship of chromosome size (log-transformed) and between-lineage FST (that is, north-south FST; Figure 5a; R2=0.84, P<0.001). Within both lineages, there was a negative relationship between nucleotide diversity and chromosome size (Figure 5b; North R2=0.48, South R2=0.41, both P<0.001). Similarly, between-lineage FST showed a strong negative relationship with nucleotide diversity for both lineages (Figure 5c; North R2=0.47, South R2=0.38, both P<0.001). For all comparisons including nucleotide diversity, the Z-chromosome (denoted as an arrow in Figure 5) appears as an outlier based on a Cook’s distance threshold of 4/(N−k−1), where N is the number of observations and k is the number of explanatory variables (Supplementary Information 3). Because the northern and southern lineage have genetic substructure within them, the above regressions were all additionally performed only including central populations in the south (E and F in Figure 1a) and western populations in the north (B, C and D in Figure 1a), which are relatively genetically unstructured based on STRUCTURE results (Figure 1b). All regressions with the reduced data sets showed similar, statistically significant results to those of the full data set (results not shown). Due to the observed relationships, we also investigated the relationship between chromosomal average recombination rates (data from Ficedula; Kawakami et al., 2014) and genetic differentiation and diversity. Here, we observed a negative relationship between recombination rates and genetic differentiation (R2=0.514, P<0.001; Figure 6) and a positive relationship between recombination rates and genetic diversity in both the northern (R2=0.507, P<0.001) and southern (R2=0.387, P<0.001) lineages.

Relationships between chromosome size, FST and nucleotide diversity (panels a–c). In (b) and (c), solid circles indicate the southern lineage and open circles indicate the northern lineage. Arrows point to the Z-chromosome in all plots. All P-values were <0.001. Only chromosomes with ⩾20 SNPs were included.

Relationship between chromosomal average recombination rate (of Ficedula flycatchers) and genetic differentiation between Certhia americana lineages. Only chromosomes with ⩾20 SNPs were included.
Discussion
Phylogeographic patterns
Next-generation sequencing has been successful in identifying fine-scale phylogeographic structure among populations in a variety of organisms, including Sarracenia pitcher plants (Zellmer et al., 2012), Ambystoma salamanders (O’Neill et al., 2012), Lycaeides butterflies (Gompert et al., 2010) and multiple species of birds (McCormack et al., 2012). These studies were generally employed when organelle DNA alone was unable to identify hypothesized phylogeographic structure. Here, we attempted to resolve discrepancies between previously-published mtDNA and nDNA phylogeographic patterns in C. americana (Manthey et al., 2011a, b). mtDNA identified three clades within the northern lineage (east, Rocky Mountains and Pacific mountain ranges), while 20 nuclear loci identified only two clades (west and east). Here, based on thousands of loci, we again recover only two genetic clusters within the northern lineage. Based on these findings, it is likely that there are large amounts of gene flow between western populations of C. americana, precluding nDNA differentiation and exhibiting patterns of mtDNA introgression that formed during periods of allopatry and subsequent contact between mtDNA lineages. In the southern lineage, we identified the same genetic clusters as the previous nDNA study (Manthey et al., 2011b), but were able to obtain strong support for lineage splitting in species tree analyses, where with only 20 loci we were unable to get strong support for relationships between all identified clades.
Within the northern and southern lineages there were differences in genetic diversity and differentiation. The southern lineage tended to have higher genetic diversity (Figure 5b) and higher average pairwise FST values (Figure 4) than the northern lineage. These differences were likely due to different patterns of within-lineage differentiation among populations, as more populations in the southern lineage are on independent evolutionary trajectories than in the northern lineage (Figure 1b).
Chromosomal diversity and differentiation
Across all well-sampled chromosomes (≥20 SNPs per chromosome), we found a strong relationship between differentiation and chromosome size (Figure 5a), assuming strong inter-chromosomal synteny in songbirds, as has been shown between songbirds with published genomes (Kawakami et al., 2014). This pattern contrasts with genomic differentiation observed between Ficedula species, which exhibit relatively stable background differentiation among chromosomes with heightened islands of differentiation on each chromosome (Ellegren et al., 2012). Although we cannot completely dismiss the possibility of genomic islands of differentiation in C. americana, if the pattern was the same as in Ficedula with one to three islands per chromosome and relatively low background divergence (Ellegren et al., 2012), we would likely expect no relationship of differentiation or diversity with chromosome size in contrast to our results in C. americana. In another species, Zonotrichia albicollis (Huynh et al., 2010), a study of many loci across chromosomes found no clear trend of differentiation between macro- and microchromosomes; this pattern may be influenced by strong selection for a chromosomal inversion mutation that is strongly linked to differential mating patterns between populations of Zonotrichia albicollis (Huynh et al., 2010). Interestingly, although there is a clear relationship between chromosome size and differentiation, this relationship is based on the log of chromosome size. When FST is plotted against raw chromosome size, the relationship is linear from 0 to 50 Mb, and plateaus in the macrochromosomes above 50 Mb. However, this relationship appears to simply be a function of fixed differences on each chromosome, as the proportion of loci with fixed differences per chromosome is strongly related with FST for each chromosome (R2=0.949, P<0.001). Alternatively, this could be explained by recombination rates, which shows a negative relationship with genetic differentiation without any transformation of the data (Figure 6).
Similar to genetic differentiation, we identified a clear relationship between genetic diversity and chromosome size in both lineages of C. americana (Figure 5b). Additionally, as would be expected due to FST being intrinsically related to within-population diversity, the relationship between genetic diversity and genetic differentiation was significant (Figure 5c); larger chromosomes showed decreased within-lineage diversity and increased between-lineage diversity (due to larger numbers of fixed differences), leading to the observed relationships of FST and chromosome size. While the Ficedula genome showed no clear pattern of diversity and chromosome length, Ellegren et al. (2012) identified reduced nucleotide diversity in almost all regions of elevated divergence.
Directional or negative selection would remove standing variation more often in larger chromosomes because the effects of selection on linked neutral polymorphisms extend further in regions of lower recombination rate (that is, smaller chromosome-wide recombination rate; Keinan and Reich, 2010); this would lead to the observed patterns in our data (Figure 5b), although our data preclude us from directly testing this relationship. Additionally, the tests for selection in BayeScan identified no outlier loci potentially under selection. Because we identified very similar genomic patterns in both lineages (that is, differentiation between and diversity within; Figure 5)—which are allopatric and geographically widespread—we find it unlikely that selective pressures would influence each lineage so similarly across such ecologically disparate regions. Here, the geographic context of genomic patterns may indicate that selection is not the dominant population genomic process influencing evolution in these lineages.
While we identified no evidence of selection, genetic drift alone may explain the pattern of the increased proportion of fixed differences and decreased genetic variability on larger chromosomes. Small C. americana effective population size would result in a tendency toward fixing genetic variation, leading to higher FST between isolated populations (in this case the northern and southern lineages) and loss of diversity within lineages. With higher relative recombination rates in smaller chromosomes, genetic diversity would be maintained relatively higher with less propensity for fixed differences, as was shown here in C. americana (Figures 3 and 5). This idea is supported by the relationships between chromosomal average recombination rates and genetic differentiation (Figure 6) and diversity.
In addition to broad chromosomal patterns, we also found that the level of differentiation between C. americana lineages on the Z chromosome was not an outlier when considered based on chromosome size (Figure 5a). This is in contrast to previous work in this species (Manthey and Spellman, 2014), as well as in contrast to other species (Carling and Brumfield, 2008, Ellegren et al., 2012), which generally show elevated differentiation in sex chromosomes compared with autosomes. Specifically, this was in stark contrast with up to 50-fold differences in levels of differentiation between the Z chromosome and autosomal background divergence levels between Ficedula species (Ellegren et al., 2012). Our previous work in C. americana (Manthey and Spellman, 2014) may have been biased in that we only used introns, compared with random coverage across the Z chromosome in this study. Alternatively, fewer autosomal fixed differences in the much smaller data set (Manthey and Spellman, 2014) may have been responsible for Z-linked markers appearing relatively more differentiated than autosomal markers. With many more loci, a more accurate picture of genomic differentiation eliminates the significance of Z-linked differentiation. In birds, most studies of differentiation between autosomes and sex chromosomes have been in closely-related hybridizing species (for example, Passerina buntings or Ficedula flycatchers; Carling and Brumfield, 2008, Ellegren et al., 2012). Perhaps a larger variety of patterns between autosomes and sex chromosomes will emerge when a more varied selection of taxonomic depths are investigated.
On Z chromosomes we also identified increased diversity, relative to chromosome size, than autosomes (Figure 5b). While lower diversity has been reported in birds’ sex chromosomes relative to autosomes, it is not a rule across species (Leffler et al., 2012). This pattern suggests background selection (that is, negative selection), and not directional selection, is acting on C. americana; as the Z chromosome is more efficient at removing deleterious mutations, deleterious alleles are maintained at relatively low frequencies with fewer neutral mutations linked to them (Begun and Whitley, 2000). This relationship should cause background selection to leave the Z chromosome more polymorphic, relative to autosomes, at neutral sites because fewer sites will be linked with a deleterious allele on sex chromosomes relative to autosomes. If directional selection was acting across C. americana, then the Z chromosome would be expected to have decreased diversity relative to autosomes (Begun and Whitley, 2000) because of lower effective population size and hemizygous expression in the heterogametic sex.
Conclusions
We sequenced thousands of SNPs, in 41 individuals of C. americana, to investigate chromosomal patterns of diversity and differentiation as well as reassess previous phyloeographic studies. Species tree analyses identified strong support for two main lineages splitting northern (United States and Canada) and southern (Mexico and Central America) populations as well as strong support for two clades within the northern lineage and three clades in the southern lineage. We identified a strong positive relationship between among-lineage genetic differentiation and chromosome size, a negative relationship between within-lineage genetic diversity and chromosome size, and a negative relationship between genetic differentiation and genetic diversity at the chromosomal level. While a combination of natural selection and genetic drift could explain these patterns, we identified no evidence of selection in the data set. Additionally, because the two geographically-broad and allopatric clades exhibited very similar genomic patterns, we find it unlikely that selective pressures for each clade would result in such similar patterns across ecologically disparate conditions. Alternatively, drift alone, leading to fixed differences between and loss of genetic variation within lineages, may explain the observed patterns. Because of relatively higher recombination rates on smaller chromosomes, larger chromosome size would, on average, lead to faster accumulation of fixed differences between (and higher FST) and loss of genetic variation within lineages, as was identified here in C. americana.
Data Archiving
All data, including SNPs, consensus sequences and associated metadata, have been deposited in the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.9h569.
References
Aguadé M, Langley CH : Polymorphism and divergence in regions of low recombination in Drosophila. (1994) Non-Neutral Evolution. Springer: USA, 67–76.
Arnold B, Corbett-Detig RB, Hartl D, Bomblies K . (2013). RADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Mol Ecol 22: 3179–3190.
Backström N, Forstmeier W, Schielzeth H, Mellenius H, Nam K, Bolund E et al. (2010). The recombination landscape of the zebra finch Taeniopygia guttata genome. Genome Res 20: 485–495.
Balakrishnan CN, Edwards SV . (2009). Nucleotide variation, linkage disequilibrium and founder-facilitated speciation in wild populations of the zebra finch (Taeniopygia guttata. Genetics 181: 645–660.
Begun DJ, Aquadro CF . (1992). Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519–520.
Begun DJ, Whitley P . (2000). Reduced X-linked nucleotide polymorphism in Drosophila simulans. PNAS 97: 5960–5965.
Bouckaert R, Heled J, Kuhnert D, Vaughan TG, Wu CH, Xie D et al. (2014). BEAST2: A software platform for Bayesian evolutionary analysis. PLoS Comp Biol 10: e1003537.
Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, RoyChoudhury A . (2012). Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol Biol Evol 29: 1917–1932.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10: 421.
Carling MD, Brumfield RT . (2008). Haldane’s rule in an avian system: using cline theory and divergence population genetics to test for differential introgression of mitochondrial, autosomal, and sex-linked loci across the Passerina bunting hybrid zone. Evolution 62: 2600–2615.
Catchen JM, Hohenlohe PA, Bassham S, Amores A, Cresko WA . (2013). Stacks: an analysis tool for population genomics. Mol Ecol 22: 3124–3140.
Ellegren H, Smeds L, Burri R, Olason PI, Backström N, Kawakami T et al. (2012). The genomic landscape of species divergence in Ficedula flycatchers. Nature 491: 756–760.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Faircloth BC, McCormack JE, Crawford NG, Harvey MG, Brumfield RT, Glenn TC . (2012). Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst Biol 61: 717–726.
Foll M, Gaggiotti OE . (2008). A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180: 977–993.
Gompert Z, Forister ML, Fordyce JA, Nice CC, Williamson RJ, Buerkle CA . (2010). Bayesian analysis of molecular variance in pyrosequences quantifies population genetic structure across the genome of Lycaeides butterflies. Mol Ecol 19: 2455–2473.
Jeffreys H . (1961) The Theory of Probability. Oxford University Press: Oxford.
Huynh L, Maney D, Thomas J . (2010). Contrasting population genetic patterns within the white-throated sparrow genome (Zonotrichia albicollis. BMC Genet 11: 96.
Kawakami T, Smeds L, Backström N, Husby A, Qvarnström A, Mugal CF et al. (2014). A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol Ecol 23: 4035–4058.
Keinan A, Reich D . (2010). Human population differentiation is strongly correlated with local recombination rate. PLoS Genet 6: e1000886.
Leffler EM, Bullaughey K, Matute DR, Meyer WK, Segurel L, Venkat A et al. (2012). Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biol 10: e1001388.
Lynch M . (2007) The Origins of Genome Architecture. Sinauer Associates: Sunderland, Massachusetts.
Manthey JD, Spellman GM . (2014). Increased differentiation and reduced gene flow in sex chromosomes relative to autosomes between lineages of the brown creeper Certhia americana. J Avian Biol 45: 149–156.
Manthey JD, Klicka J, Spellman GM . (2011a). Isolation-driven divergence: speciation in a widespread North American songbird (Aves: Certhiidae). Mol Ecol 20: 4371–4384.
Manthey JD, Klicka J, Spellman GM . (2011b). Cryptic diversity in a widespread North American songbird: Phylogeography of the Brown Creeper (Certhia americana. Mol Phylogenet Evol 58: 502–512.
McCormack JE, Maley JM, Hird SM, Derryberry EP, Graves GR, Brumfield RT . (2012). Next-generation sequencing reveals phylogeographic structure and a species tree for recent bird divergences. Mol Phylogenet Evol 62: 397–406.
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA . (2007). Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res 17: 240–248.
Mugal CF, Nabholz B, Ellegren H . (2013). Genome-wide analysis in chicken reveals that local levels of genetic diversity are mainly governed by the rate of recombination. BMC Genomics 14: 86.
Nachman MW . (2001). Single nucleotide polymorphisms and recombination rate in humans. Trends Genet 17: 481–485.
Nylander JA, Wilgenbusch JC, Warren DL, Swofford DL . (2008). AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 24: 581–583.
O’Neill EM, Schwartz R, Bullock CT, Williams JS, Shaffer HB, Aguilar-Miguel X et al. (2012). Parallel tagged amplicon sequencing reveals major lineages and phylogenetic structure in the North American tiger salamander (Ambystoma tigrinum species complex. Mol Ecol 22: 111–129.
Parchman TL, Gompert Z, Mudge J, Schilkey FD, Benkman CW, Buerkle C . (2012). Genome-wide association genetics of an adaptive trait in lodgepole pine. Mol Ecol 21: 2991–3005.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
R Development Core Team. (2012) R: A Language for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria.
Rambaut A, Drummond AJ . (2007). TRACER v1.4. Available at beast.bio.ed.ac.uk/tracer.
Rheindt FE, Fujita MK, Wilton PR, Edwards SV . (2013). Introgression and phenotypic assimilation in Zimmerius flycatchers (Tyrannidae): population genetic and phylogenetic inferences from genome-wide SNPs. Syst Biol 63: 134–152.
Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Künstner A et al. (2010). The genome of a songbird. Nature 464: 757–762.
Zellmer AJ, Hanes MM, Hird SM, Carstens BC . (2012). Deep phylogeographic structure and environmental differentiation in the carnivorous plant Sarracenia alata. Syst Biol 61: 763–777.
Acknowledgements
We thank Lindsey Allen and Oxana Gorbatenko for assistance in the lab. Scott V Edwards and Simon Malcomber provided useful feedback on earlier drafts of this manuscript. This work was supported in part by NSF grants (DEB 0815705) to JK and (DEB 0814841) to GMS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article

Cite this article
Manthey, J., Klicka, J. & Spellman, G. Chromosomal patterns of diversity and differentiation in creepers: a next-gen phylogeographic investigation of Certhia americana. Heredity 115, 165–172 (2015). https://doi.org/10.1038/hdy.2015.27
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.27
This article is cited by
-
Gene flow and genetic admixture across a secondary contact zone between two divergent lineages of the Eurasian Green Woodpecker Picus viridis
Journal of Ornithology (2019)
-
Genomic insights into hybridization in a localized region of sympatry between pewee sister species (Contopus sordidulus × C. virens) and their chromosomal patterns of differentiation
Avian Research (2016)
-
Conservation genomics reveals multiple evolutionary units within Bell’s Vireo (Vireo bellii)
Conservation Genetics (2016)
