
Abstract
It is commonly found that individual hybrid, polyploid species originate recurrently and that many polyploid species originated relatively recently. It has been previously hypothesized that the extremely rare allopolyploid peat moss Sphagnum troendelagicum has originated multiple times, possibly after the last glacial maximum in Scandinavia. This conclusion was based on low linkage disequilibrium in anonymous genetic markers within natural populations, in which sexual reproduction has never been observed. Here we employ microsatellite markers and chloroplast DNA (cpDNA)-encoded trnG sequence data to test hypotheses concerning the origin and evolution of this species. We find that S. tenellum is the maternal progenitor and S. balticum is the paternal progenitor of S. troendelagicum. Using various Bayesian approaches, we estimate that S. troendelagicum originated before the Holocene but not before c. 80 000 years ago (median expected time since speciation 40 000 years before present). The observed lack of complete linkage disequilibrium in the genome of this species suggests cryptic sexual reproduction and recombination. Several lines of evidence suggest multiple origins for S. troendelagicum, but a single origin is supported by approximate Bayesian computation analyses. We hypothesize that S. troendelagicum originated in a peat-dominated refugium before last glacial maximum, and subsequently immigrated to central Norway by means of spore flow during the last thousands of years.
Similar content being viewed by others

Introduction
Sympatric speciation can occur by interspecific hybridization associated with genome doubling, giving rise to allopolyploid daughter species. There is a broad consensus that this is an important mode of speciation in a range of organisms, plants in particular (Mallet, 2007; Soltis and Soltis, 2009). Although most or possibly all plant species may have polyploidization at some time in their ancestry (Soltis and Soltis, 2009; Wood et al., 2009), the proportion of angiosperm and fern speciation events accompanied by ploidal increase has been estimated at 15 and 31%, respectively (Wood et al., 2009). The proportion of polyploids has been estimated as somewhat lower for mosses (5–10%) and liverworts (6–19%), and these appear to be mainly allopolyploids (Såstad, 2004, but see Shaw (2009)). Many studies have shown that genome doubling may be accompanied by massive genome alterations, including loss of DNA and changes in gene expression (for example, Adams, 2007; Chen, 2007; Gaeta and Chris Pires, 2009).
Hybridization has several evolutionary consequences, including increased intraspecific genetic diversity and transfer of genetic adaptations between taxa (Martin et al., 2006; Whithney et al., 2006; Castric et al., 2008), origin of new ecotypes and species, as well as reinforcement or breakdown of reproductive barriers (reviewed by Rieseberg (1997) and Rieseberg and Willis (2007)). Furthermore, it is widely recognized that recurrent origins is the rule, not the exception for most polyploid species (Soltis and Soltis, 1999; Soltis et al., 2003; Abbott and Lowe, 2004; Shaw, 2009). Recurrent polyploidization involving genetically different diploids can create a series of genetically distinct polyploid populations, and gene flow between polyploid populations of independent origin permits recombination and production of additional genotypes (Soltis and Soltis, 1999). Recurrent formation may be critical to counterbalance local extinction of small populations (Soltis et al., 2003), and this may be one reason why there are few examples of extant polyploid taxa of vascular plants with single origins. Several vascular plant species have been shown not only to have originated multiple times, but also to have originated very recently, within hundreds or a few thousands of years (Abbott and Lowe, 2004; Soltis et al., 2004; reviewed by Soltis and Soltis (2009)).
Sphagnum troendelagicum Flatb. (Flatberg, 1988) has been shown to be an allopolyploid derivative of S. balticum (Russ.) C. Jens and S. tenellum (Brid.) Brid. (Såstad et al., 2001). S. troendelagicum is only known from five proximate areas in oceanic, boreal parts of central Norway (Flatberg, 1988), whereas the parental species are widespread throughout the Northern Hemisphere (Daniels and Eddy, 1985). It is one of the very few Norwegian endemic species, and it is classified as critically endangered (Flatberg et al., 2006a, 2006b). On the basis of studies of natural populations using random amplified polymorphic DNA (RAPD) markers (Williams et al., 1990, Stenøien and Flatberg (2000) hypothesized that S. troendelagicum may have originated multiple times. The main reason for this conclusion was the finding of relatively low linkage disequilibrium (LD) even though no sporophytes (that is, sexual reproduction) have ever been observed for these plants in the field. Still today, no sporophytes have been observed in natural populations of this species (KI Flatberg, personal observation). In addition, the total amount of genetic variability within S. troendelagicum is quite high even though local population sizes are small. On the basis of these observations, and given what we know about plant polyploidization (Soltis and Soltis, 1993; Soltis et al., 2004), one can speculate that S. troendelagicum is a young species possibly having originated several times after the last ice age c. 11 000 years before present (bp), in or very close to localities where the plants are found today. Both parental species occur sympatrically with S. troendelagicum at most sites where it is found.
It has been shown that RAPD markers may have low reproducibility (Pérez et al., 1998; Rabouam et al., 1999, but see Ramos et al. (2008)), and previous results concerning levels of variability and inferred recombination in S. troendelagicum are therefore difficult to interpret with confidence. The aim of this study is to learn more about the evolutionary history of this species, more specifically to (1) determine which of the parental taxa are maternal and paternal progenitors, respectively; (2) quantify patterns of genetic variability and structuring in natural populations of S. troendelagicum and the parental species using variable markers with high reproducibility; (3) test the hypothesis that the species originated multiple times; (4) quantify historical parameters, most importantly time since speciation event(s), to determine whether the species likely originated in central Norway or whether it immigrated after the last ice age; and finally, (5) make suggestions concerning the continued preservation of S. troendelagicum.
Materials and methods
Sampling of material
Sphagnum troendelagicum, S. balticum and S. tenellum were sampled from mire localities at four of the five known areas it has been found, viz. Fosnes, Grong, Høylandet and Overhalla municipalities in the northern parts of central Norway (Supplementary Table S1). S. balticum and S. tenellum were also sampled from six mire localities outside the known distribution area of S. troendelagicum in more southern parts of central Norway. As far as possible, a minimum of 10 population samples of each species were sampled at each locality, mostly with a minimum distance of 10 m between samples. More samples were included from the Grong, Høylandet and Overhalla localities, particularly of S. troendelagicum, but in Fosnes and one of the localities from Selbu (S Haukåtjønna) only five or six samples were made because of smaller total population sizes. The samples were air dried and stored in paper bags at room temperature. In addition, a number of selected herbarium specimens of the three species from Norway, Finland, Japan and North America were also included in the study (Supplementary Table S1).
Molecular analyses
Total DNA was extracted from dry capitulum tissue from each sample using the SP Plant DNA Mini Kit and the Plant DNA Kit (OMEGA bio-tek, http://www.omegabiotek.com) according to the manufacturer's protocols. Individuals were genotyped at 12 microsatellite loci previously described for Sphagnum (Shaw et al., 2008b), that is, markers 4, 9, 10, 14, 17, 18, 19, 20, 22, 28, 29 and 30. Vouchers for each population, including the whole samples and the remaining part of the plants from which the DNA is extracted from are deposited at the herbarium in Trondheim (TRH). DNA and tissue material from S. troendelagicum is also deposited in the ColdGene genome resource bank (http://www.vm.ntnu.no/coldgene_en/).
Thirteen samples of S. troendelagicum in addition to 12 samples of S. balticum and 16 samples of S. tenellum from across the species distribution ranges (Supplementary Table S1) were sequenced for the chloroplast area trnG (for primer information, see Pacak and Szweykowaska-Kulinska, 2000). PCR amplification and sequencing were accomplished using protocols described in Shaw et al. (2003).
Population genetic analyses
Overall summary statistics were estimated for DNA sequence and microsatellite data. For the trnG data, we estimated several within- and between-group statistics, including numbers of segregating sites (S), numbers of mutations, haplotypic diversity and nucleotide diversity (π) within and between taxa using the DnaSP 5.00.07 software (Librado and Rozas, 2009). Microsatellite diversity was assessed by calculating the proportion of polymorphic loci, mean number of pairwise differences between individuals and average gene diversity over loci (HE; Nei, 1987). The presence of LD between all pairs of loci was tested for all three species using an exact test of LD (Slatkin, 1994). For S. troendelagicum, the ‘gametic disequilibrium’ was estimated based on genotypes instead of alleles without them needing to specify gametic phase. Loci with missing alleles were excluded from LD analyses to obtain maximally conservative estimates. The percentage of linked loci in relation to the maximum number of linked loci (Pd) was estimated for each population as described in Stenøien and Såstad (1999). We estimated fixation indices, FST, among S. troendelagicum populations, and the corrected numbers of pairwise differences between S. troendelagicum populations, and between S. troendelagicum and parental species populations (Weir and Cockerham, 1984; Michalakis and Excoffier, 1996). The significance of differentiation measures was tested using the non-parametric permutation approach described by Excoffier et al. (1992). Overall genetic diversity, LD and genetic structure were analysed with the aid of the software Arlequin ver. 3.0 (Excoffier et al., 2005).
To further explore genetic structuring of the three species, we performed Bayesian cluster analysis on genotypic data using the program Structure ver. 2.3.1 (Pritchard et al., 2000). We evaluated the most likely number of clusters (K) by aid of the ΔK statistics (Evanno et al., 2005). We assumed an admixture model with independent allele frequencies with sampling locations as prior information to assist in the clustering (Hubisz et al., 2009). Ten runs were performed for each K from 1 to 15, with 100 000 steps of burn-in and 100 000 steps per run.
Hypothesis testing
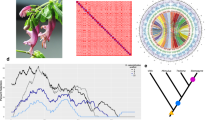
We wanted to test the hypothesis that S. troendelagicum originated once, twice or four times for the four populations investigated (that is, for Fosnes, Grong, Høylandet and Overhalla), and this was carried out in a coalescence framework using approximate Bayesian computation (ABC; Beaumont et al., 2002). The three historical scenarios evaluated are presented in Figures 1a–c. In scenario 1 (Figure 1a), it is assumed that S. troendelagicum originated only once by hybridization between S. balticum and S. tenellum t4 years in the past. After that, the S. troendelagicum lineage split three times to form the four populations investigated in this study; that is, at t3, t2 and t1 years in the past, respectively. S. balticum and S. tenellum diverged from each other tA years in the past. It is assumed that the effective population sizes of the two parental species have been similar and constant through time (NA). It is also assumed that the four S. troendelagicum populations have had similar effective population sizes through their histories (Nt). S. troendelagicum consists of 50% S. balticum alleles and 50% S. tenellum alleles found in the parental species populations at time t4. For a relatively short period after this speciation event (equal to x years), the effective population size of the S. troendelagicum population (N0) is assumed to be low. Note that time is not to scale in Figure 1, and also that even though t4 must have occurred before t3, there are no assumptions in the model with regard to the relative order of t2 and t1 (see below for an overview of different assumptions in the models).

Three scenarios for the evolution of S. troendelagicum populations were studied here, the major difference being the number of times the species has independently originated (once under scenario 1 (a), twice under scenario 2 (b) and four times under scenario 3 (c); see text above for details). The relative branching in scenarios 1 and 2, that is, the assumption that Overhalla and Høylandet are more closely related than Fosnes and Grong, are based on results of the Bayesian cluster analyses.
In scenario 2 (Figure 1b), S. troendelagicum originated twice, first at time t4 and second (independently) at time t3. After that, the two S. troendelagicum lineages split at times t2 and t1, respectively, to form the four populations. Again, the parental species split at tA, 50% of each of the parental genomes forms the initial S. troendelagicum populations at t4 and t3, and population sizes of the newly formed S. troendelagicum populations are N0 in a period of x years after the speciation events. There are no assumptions with regard to the relative order of t3 and t4, but t4>t2 and t3>t1.
Finally, in scenario 3 (Figure 1c), all S. troendelagicum populations originated independently of one another at times t1–t4 in the past. Moreover, here each speciation event is associated with the parameters x and N0 as above, each parental species contributed 50% of the genetic material to the newly formed populations and the relative orders of t1–t4 are not determined a priori. The parental species are coded as diploids in the input data set in these analyses, and all the populations of each parental species have been grouped together in single global populations.
Several additional assumptions are included in the models. First, the times for the various events (that is, the various t's) in the three scenarios are not dependent on one another. For example, t4 in scenario 1 is calculated independently from t4 in scenarios 2 and 3 (technically, t4 is treated as separate parameters t5 and t7 in scenarios 2 and 3, respectively, and for technical reasons the same approach is followed for other parameters). Second, it is assumed that tA must be greater than t1, t2, t3 and t4, that t4>t3 and t3 is larger than both t2 and t1 in scenario 1 and that t4>t2 and t3>t1 in scenario 2. Previous information about the parameters are given, with NA and Nt limits set to 1000–1 000 000, whereas N0 limits set to 100–100 000, the events t1–t4 are occurring between 1000–1 000 000 years ago, while tA occurred 10 000–10 000 000 years ago, and x lasted between 1 and 1000 years. We assumed a stepwise mutation model (Ohta and Kimura, 1973) with no indel mutations. In plants, microsatellite mutation rates have been found to vary widely (for example, 1 × 10−2 in chickpeas (Udupa and Baum, 2001) and 1 × 10−6 in Dictyostelium discoideum (McConnell et al., 2007). Previous information on mutation rates will highly influence estimated divergence times. On the basis of previous estimates of microsatellite mutation rates varying between 10−6 and 10−4 per year in S. fimbriatum (Szövényi et al., 2008), we used 10−5 as an average mutation rate per year for the 12 simulated loci. Each locus were allowed to mutate to a number of repeats equal to the maximum observed numbers for each of the loci; thus, 40, 165, 104, 92, 134, 44, 56, 101, 77, 54, 94, 117 repeats for loci 22, 28, 4, 9, 10, 18, 17, 20, 29, 30, 19, 14, respectively (Shaw et al., 2008b). In each simulation, the populations are sampled at t0 and summary statistics are computed for each sample; that is, mean number of alleles, mean genetic diversity, mean size variance for each individual sample, mean M index (ratio of number of alleles to range size) across loci (Garza and Williamson, 2001; Excoffier et al., 2005) and mean index of two-sample classification (Rannala and Mountain, 1997; Pascual et al., 2007). Data sets (500 000) were generated for each scenario; that is, 1 500 000 simulations were performed in total. The scenarios were compared using two approaches: one by directly comparing the summary statistics with the observed diversity in the data set and counting the frequency of the various scenarios among the most similar simulated data sets (direct estimate approach; Miller et al., 2005; Pascual et al., 2007), and one by doing a logistic regression of each scenario probability for the most similar simulated data sets on the deviations between simulated and observed summary statistics (Fagundes et al., 2007; Beaumont, 2008). In these two comparisons, 0.1 and 1% simulated data closest to the observed values were used, respectively. The ABC computations and parameter estimations were performed using the DIY-ABC ver. 0.72 software (Cornuet et al., 2008).
Estimation of historical parameters
Two approaches were used to estimate historical parameters. First, the 1% simulated data sets from the ABC analyses (see above) that fit observed data most closely were used to inform the posterior distribution of parameter values (that is, t1–t4, tA, NA, N0, Nt and x) using logit-transformation of original and composite parameter values (Beaumont et al., 2002). As suggested previously (Excoffier et al., 2005), the reference tables were used together with pseudo-observed data having known parameter values to compute estimation bias of parameter estimates. The average relative bias was used for this, defined as the difference between estimates and true values averaged over all test data sets. One per cent of simulated data sets was used in the evaluation, with logit-transformed data and fixed parameter values. Second, the isolation-with-migration (IM) model was used to estimate time since divergence (that is, allopolyploidization) between S. troendelagicum and each of the parental species using the IM software (Nielsen and Wakeley, 2001; Hey and Nielsen, 2004). The IM model assumes an ancestral population being split into two daughter populations at some time while allowing subsequent migration between these two populations. Coalescence simulations are used to determine maximum-likelihood parameter estimates given the genetic data and prior information. This model is not ideal for exploring time since polyploidization events, because two populations are merged rather than one population being split into two. To use this approach, it is necessary to split the S. troendelagicum genome into ‘tenellum’ and ‘balticum’ alleles for the various loci (that is, assess from which parental species the alleles have been derived), and then use this information in two IM analyses: one involving S. tenellum and one involving S. balticum. It was possible to use six loci in the comparison between S. troendelagicum and S. balticum, in which it was clear that alleles in S. troendelagicum must have been derived from S. balticum. In the comparison involving S. tenellum, only four loci could be used. Only individuals containing no missing values for any of these loci were used for any of the data sets. Three independent IM runs were performed on each of the data sets (six runs in total), with generation time arbitrarily set to 5 years. Generation times in peat mosses may be larger than 5 years (for example, Flatberg et al., 2006a, 2006b), but this prior value has no influence on divergence time estimates. In the IM runs, we assumed no subsequent migration after speciation (that is, divergence). This latter approach would tend to underestimate divergence time if subsequent introgression has occurred to any significant extent. It must also be assumed that only one speciation event has occurred in the history of the sample. We used 10−5 as an average mutation rate per year in the IM analyses (Szövényi et al., 2008). The two latter runs for each species pair was set to a minimum of 61 million steps after burn-in, with effective sample size (ESS) values being at least 600.
Results
Data considerations
In total, 326 individuals were genotyped (126 S. tenellum, 86 S. balticum and 114 S. troendelagicum). A high number of S. troendelagicum individuals exhibited one or several homozygous genotypes for individual loci, or missing data, and a few individuals of the parental taxa exhibited single heterozygous loci. Only two chromosome numbers, N=19 and 38, have been reported in Sphagnum (Fritsch, 1991). On the basis of chromosome counts, flow cytometric estimates of genome sizes, isozyme patterns and microsatellite allelic profiles, S. balticum and S. tenellum are known to have haploid gametophytes (that is, N=19), whereas S. troendelagicum has diploid gametophytes (Såstad et al., 2001). As allopolyploid origin and fixed heterozygosity have been shown for isozyme markers (Såstad et al., 2001), we have chosen to adopt a conservative strategy by treating one allele as missing data in homozygous S. troendelagicum if the parental species do not possess the same allele. Similarly, we deleted one allele in heterozygous S. balticum and S. tenellum loci, using the most common allele in the population in further analyses. We observe relatively high number of missing data, and inferred levels of variability should thus be interpreted with care.
Genetic variability
Forty-one chloroplast DNA (cpDNA; trnG) sequences are deposited to GenBank (accession numbers HM439652-HM439692), and results from genetic analyses are presented in Tables 1, 2 and 3. The only haplotype found among S. troendelagicum individuals is identical to the most common haplotype found in S. tenellum (Table 1). DNA sequence diversities are presented in Table 2. S. troendelagicum is monomorphic in the trnG region, whereas S. balticum is more variable than S. tenellum for various diversity statistics, including number of segregating sites (16 versus 1, respectively) and nucleotide diversity (π±s.d. equal to 0.0066±0.0028 and 0.0002±0.0001, respectively). Levels of differentiation between the three species for the trnG region are summarized in Table 3. S. troendelagicum and S. tenellum are not differentiated to any significant extent, whereas S. balticum is more differentiated from both of the two other species with four fixed nucleotide differences between S. balticum and both S. tenellum and S. troendelagicum.
In the microsatellite analyses, loci with less than 50% missing data were included in the analyses of genetic diversity; levels of genetic variability within populations of the three species are presented in Table 4. Genetic variability is found in parental species populations, with an average 4.4 and 3.0 polymorphic loci per population of S. balticum and S. tenellum, respectively, implying an average proportion of polymorphic loci of 51 and 41%, respectively. Average HE is 0.18 (±0.04 s.e.) and 0.11 (±0.02 s.e.) per population in S. balticum and S. tenellum, respectively, whereas the mean number of pairwise differences is 1.46 (±0.38 s.e.) and 0.88 (±0.21 s.e.), respectively. Levels of LD are low in populations of the parental species (average Pd values found to be 7 and 6% in S. balticum and S. tenellum, respectively). Levels of genetic variability are high in diploid S. troendelagicum compared with the parental species because of fixed heterozygosities. On average, 10.8 polymorphic loci occur per population, yielding an average proportion of polymorphic loci of 69%. Expected HE is 0.34 (±0.20 s.e.) and mean pairwise differences are 3.99 (±0.22 s.e.) per population. Levels of LD are less than possible maxima in S. troendelagicum, with Pd values ranging from 47 to 100% within populations (66% on average), and an average Pd for the whole species is 43%.
Genetic structure
There is significant genetic differentiation among S. troendelagicum populations (FST ranging from 0.12 to 0.16; Table 5). Average FST values between S. troendelagicum and the parental species are 0.09 to S. balticum and 0.19 to S. tenellum, whereas FST between S. balticum and S. tenellum is 0.46 (all P-values <0.001). Thus, FST values between S. troendelagicum and each of the parental species are relatively similar to FST values estimated between S. troendelagicum populations. In Table 6, corrected numbers of pairwise differences between individuals in different S. troendelagicum populations and local populations of the parental species are presented, as well as the average pairwise differences between S. troendelagicum and parental species populations. S. troendelagicum populations are not more similar to local S. balticum populations than to other populations of this species. However, there is a tendency for S. troendelagicum populations to be more similar to local S. tenellum populations than to other populations of S. tenellum.
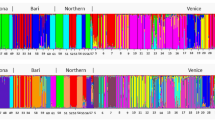
In the Bayesian cluster analysis, ΔK had the highest estimated value for K=2 (Figure 2). When K=2, S. balticum populations are mostly assigned to one group, all S. tenellum populations are mostly assigned to the second group and all S. troendelagicum populations are assigned to both the groups (approximately 40–60% to each group for each S. troendelagicum population; Figure 3). At K=3, the S. troendelagicum populations are divided into two groups: Fosnes and Grong to one, and Høylandet and Overhalla to the other. Some populations of the parental species are also assigned to this third group, in particular S. tenellum individuals at Grong (results not shown). At K=4, both S. troendelagicum and S. balticum populations are separated into two major groups each (Figure 4).

Results of Structure analyses, where probabilities of different K values (LnP(D); red line) and ΔK values (blue line) are plotted against values of K from 1 to 10 (results from K values up to 15 not shown). LnP(D) increases for increasing values of K and it is hard to determine the exact breaking point. ΔK on the other hand clearly peaks at K=2.

Results from clustering of individuals of the three species at the most probable value of K (K=2), as measured by the delta K statistics. S. balticum populations are mostly assigned to cluster 1 (91–99%), S. tenellum populations are mostly assigned to cluster 2 (90–100%, except Grong and Overhalla with 80 and 82%, respectively) and S. troendelagicum populations assigned approximately one-half to each of the clusters (at least 41% assigned to one of the clusters).

Results of Structure analyses run for the different S. balticum, S. tenellum and S. troendelagicum populations when K=4 clusters are considered. For each locality, S. balticum is presented first, followed by S. tenellum and third by S. troendelagicum at Fosnes, Høylandet, Overhalla and Grong localities. S. troendelagicum populations can be separated into two main groups (Fosnes-Grong versus Høylandet-Overhalla), and S. troendelagicum and S. tenellum are genetically similar at the Grong locality.
Number of independent origins of S. troendelagicum
In the ABC analyses, the numbers of simulated data sets with summary statistics similar to observed values based on the various scenarios are treated as posterior probabilities for the three scenarios. The direct approach does not reveal clearcut differences in probabilities of the three scenarios (results not shown). The logistic regression approach supports scenario 1, that is, a single origin of S. troendelagicum (P>0.9846).
Estimation of historical parameters
In the ABC approach, simulations based on scenario 1 with summary statistics close to observed values were used to estimate parameters included in the model. Results from these calculations are presented in Table 7. The parental species seem to have diverged approximately 270 000 years bp (tA, median value). Around 40 000 years ago (t4), S. troendelagicum was formed by hybridization between these two diverging species. In a period after this initial formation, most likely ∼450 years (x), the effective size of this newly formed species, N0, was ∼50 000, and long-term effective population size (Nt) is >110 000. The effective sizes of the parental species are approximately >60 000 individuals each. At approximately 11 000 years bp, the species split into two major lineages (t3), and at 900 years bp (t2), the Høylandet and Overhalla populations split, whereas somewhat later, at 600 years bp (t1), the Fosnes and Grong populations split.
The mean relative bias varied among parameters (Table 8), with extreme median values ranging from 0.03 (NA) to 3.85 (t1) and 63.83 (N0). All other median values were in the range 0.15–1.49, with t4 having a median mean relative bias of 0.37. This means that an estimated t4 of 40 000 years implies that the true value could be wrong by approximately 10 000 years, and one can speculate that 30 000 years may be a better estimate of time since origin.
The results from the IM analyses are summarized in Figure 5, with divergence time between S. troendelagicum and S. balticum having a highest probability for t=14 200 years bp (averaged over two long runs, 95% confidence interval between 4750 and 73 850), and divergence time between S. troendelagicum and S. tenellum having a highest probability for t=21 000 years bp (also averaged over two long runs, 95% confidence interval between 6650 and 85 750). Divergence times before the last glacial maximum and up to 40 000 years bp thus have relatively high probabilities in both IM analyses. On the basis of the combined ABC and IM results, although, it seems unlikely that S. troendelagicum is younger than the last glacial maximum (c. 11 600 years) or older than 80 000 years.

Results of IM runs comparing divergence between S. troendelagicum and S. balticum (red lines) and S. balticum (blue lines), respectively. Results from two runs for each species are presented (represented with different colours). Time since divergence is measured in years before present, and the y-axis represents likelihood of the estimated distribution.
Discussion
Stenøien and Flatberg (2000) reported relatively high genetic variability within S. troendelagicum compared with other studied peat mosses (for example, gene diversity comparable with levels found in the common circumboreal S. fallax; Stenøien and Såstad, 1999). They also found very low levels of LD (average Pd equal to 4.8%) and low differentiation between S. troendelagicum populations (average FST equal to 0.07). The high variability and low LD in an apparently asexual species led the authors to suggest that S. troendelagicum may have originated several times, possibly at each locality it is found presently. This latter would imply very recent speciation events, as current central Norwegian peatlands may have originated 5000–8000 years bp (Solem, 1986). However, a scenario involving multiple origins, perhaps one origin at each local site, should yield high genetic differentiation among populations if there is limited gene flow. A peat moss without sexual reproduction and spore production should have an extremely limited gene flow, even though limited dispersal by vegetative fragments could in theory be possible over short distances (Stenøien and Såstad, 1999). The results reported by Stenøien and Flatberg (2000) could therefore perhaps equally likely fit a scenario of a single origin of S. troendelagicum, given sufficient time since origin for the accumulation of genetic variation by mutations, some sexual reproduction and some gene flow between populations. However, distinguishing between these different scenarios becomes problematic since the reproducibility of the anonymous genetic markers (RAPDs) employed by Stenøien and Flatberg (2000) is uncertain.
In this study, we find higher levels of within-population variability than reported earlier. Average frequencies of polymorphic loci are almost five times higher in microsatellites compared with RAPDs, and average gene diversity is approximately 10 times higher for the Fosnes and Høylandet S. troendelagicum populations, and >500 times higher for the Overhalla population compared with earlier reports (Stenøien and Flatberg, 2000). The levels of genetic variability within populations are comparable with the selfing model angiosperm Arabidopsis thaliana (for example, Stenøien et al., 2005). This means that S. troendelagicum harbours appreciable levels of genetic variability, even though the absolute levels are not very high compared with widespread outcrossing species, as pointed out also by Stenøien and Flatberg (2000). Interestingly, the level of microsatellite variability is much lower than found in S. beringiense, another peat moss not yet found with sporophytes (Shaw et al., 2008a). It seems that mating system is not a major determinant on levels of genetic variability in many bryophytes (see Stenøien and Såstad, 2001). Similar cpDNA haplotypes are found in S. troendelagicum and S. tenellum, and as cpDNA is maternally inherited in Sphagnum (Natcheva and Cronberg, 2007), our results suggest that S. tenellum is the maternal species in the hybridization event or events leading to the present S. troendelagicum populations.
Boatman and Lark (1971) hypothesized that sexual reproduction may be of limited ecological importance in Sphagnum, as establishment from spores have never been observed in the field, and as spores are hard to germinate in mire water. We are interested in judging whether patterns of genetic variability in S. troendelagicum are due to sexual recombination, multiple origins or both. Our results concerning LD and inferred recombination in S. troendelagicum must be interpreted with caution because polymorphisms are often represented as relatively few individuals with low-frequency genotypes, making it difficult to perform statistical tests of association. Our results do, however, support earlier claims that either recombination must have occurred in the history of S. troendelagicum, or the species must have been formed multiple times. This is evidenced by the observation that only about 66% of maximum LD is encountered in sampled microsatellite loci. Such levels are frequently found even in populations of the highly selfing model angiosperm Arabidopsis thaliana (Lundemo et al., 2009) and does not necessarily imply very recent or frequent recombination within populations.
We believe that the low levels of LD reflect recombination rather than multiple speciation events for two reasons. First, in mosses dispersal occurs most effectively via spores (Van Zanten, 1978; Longton, 1997; Sundberg, 2005), and even movement between local populations in central Norway is difficult to imagine without the aid of spores. Second, lack of LD at the species level can be explained by recurrent speciation, but one would then expect complete or almost complete LD within populations if migration is limited between populations due to lack of spore-mediated gene flow. This is not the case in the present populations, as Pd values are relatively low also within populations. We therefore conclude that sexual recombination has occurred repeatedly throughout history of S. troendelagicum. This does not preclude the possibility that this species may have originated multiple times, though.
This study reveals somewhat higher genetic structuring than that reported earlier, with average FST values being twice as high for microsatellites compared with RAPD markers (FST being on average equal to 0.14 in this study compared with 0.07 in Stenøien and Flatberg (2000)). The differentiation between S. troendelagicum populations is approximately the same as the differentiation between S. troendelagicum and each of the parental species. S. troendelagicum populations are not more similar to local S. balticum populations than other populations of this species. However, there is a tendency for S. troendelagicum populations to be more similar to local S. tenellum populations than other populations of S. tenellum (Table 6), especially at the Grong locality (Figure 4). This could be so because S. troendelagicum at least at some sites has been formed by the local populations of the maternal species. Alternatively, one cannot rule out the possibility that backcrossing between S. troendelagicum and S. tenellum has taken place at these sites. Such backcrossing between an allopolyploid and one of its parents was documented by Flatberg et al. (2006a, 2006b) for species in Sphagnum section Acutifolia and by Ricca (unpublished) for species in section Subsecunda.
Interestingly, cluster analysis reveals two main clusters in the total microsatellite data set, with the parental species largely belonging to one of the clusters each and S. troendelagicum individuals admixed. It is clear from Figure 3 that S. troendelagicum populations group into two subgroups, with Fosnes and Grong forming one subgroup and Høylandet and Overhalla forming the other. This is even clearer for the results of the cluster analysis resolving four main clusters (Figure 4). The parental species are clearly genetically differentiated at each of the localities where they co-occur and where S. troendelagicum is not found. On localities where S. troendelagicum is also found, the parental species are also genetically dissimilar, suggesting that hybridization is not regularly occurring at these sites either. Nevertheless, the two subgroups found in S. troendelagicum may suggest at least two independent speciation events. There are no indications of S. troendelagicum having immigrated to the Trøndelag region from other parts of the world (that is, North America or Asia) at some time in the past, as parental species genotypes from these localities are not genetically more similar to S. troendelagicum than those found within the Trøndelag region.
From the analyses presented above, it is possible to formulate various hypotheses concerning the evolutionary history of S. troendelagicum. First, it seems reasonable to assume that it is a fairly young species; this is supported by the relatively low genetic divergence between S. troendelagicum and the parental species. Furthermore, the lack of cpDNA nucleotide differences between S. troendelagicum and the maternal progenitor species, S. tenellum, supports a hypothesis of relatively recent speciation. It must be remembered, though, that for a sequence of 1000 years bp (approximate size of trnG) with a substitution rate of up to 3 × 10−9 per nucleotide site per year (Wolfe et al., 1987), one would not expect an interspecific substitution to take place before 3 million years after divergence. In addition, the conservative approach taken when scoring microsatellite data will tend to underestimate genetic structuring and levels of genetic variability, and one cannot rule out the possibility that populations contain more variability than what we document based on our approach.
Second, it is clear that S. troendelagicum populations are grouped into two major clusters (Fosnes and Grong in one cluster, and Høylandet and Overhalla in the other cluster). This is not easily reconciled with a hypothesis of this species being formed multiple times at local sites by hybridization of S. balticum and S. tenellum populations located there. This latter scenario would perhaps imply stronger genetic structuring between S. troendelagicum populations found at the four localities, and it would also imply that one should find lower genetic differentiation between S. troendelagicum and the parental species at local sites compared with parental species’ populations found elsewhere. As mentioned, measures of pairwise population differentiation do not indicate that local S. balticum individuals have been involved in the formation of local S. troendelagicum populations, even though our results do not strongly contradict this possibility. It cannot be ruled out that local S. tenellum has been involved in such multiple speciation events. In any case, the presence of two S. troendelagicum clusters raises the question as to whether the species has been formed by at least two independent hybridization events.
Here we tested the three scenarios presented in Figure 1 using the ABC approach (Beaumont et al., 2002), and based on these analyses we can conclude that the present data set support a hypothesis of one single origin for S. troendelagicum (Figure 1a). The assumptions in this test are violated for polyploids (see below), and the results must therefore be interpreted with caution. Nevertheless, a single origin is not an implausible scenario since there is no strong genetic structuring observed between S. troendelagicum populations. Such structuring would have been expected if different populations had separate evolutionary histories. If different parental genotypes had contributed to different S. troendelagicum clades, then the presence of different alleles in various clades would yield FST's of 1 between them. Nevertheless, if extensive gene flow has occurred between the populations throughout history, then any signal of independent origins may have been lost. We can therefore not entirely rule out the possibility that S. troendelagicum has originated several times. What we can conclude is that if we consider one speciation event as the null hypothesis, our data cannot reject this hypothesis.
The IM and ABC approaches yield different estimates for the time since origin of S. troendelagicum. This is as expected given the differences in assumptions in the two models, as well as the differences in data manipulations previous to the analyses. The stringent criteria concerning choice of homologous alleles to be included in the IM analyses most certainly pushes divergence time estimates down to the lower end of the scale, and IM results should therefore be viewed as minimum estimates of divergence. It is interesting to note that the estimated time since divergence between S. troendelagicum and the two parental taxa are quite similar, though. In the ABC analyses, one uses a coalescence approach based on the Wright–Fisher model in the simulation of gene trees to be compared with the real data set. In this model, alleles are in each generation drawn from a gamete pool of infinite size and assembled into genotypes to form the next generation. The assumptions of the model are not fulfilled in an allodiploid species lacking free recombination, as alleles are paired in the gametes, that is, a random draw of one allele causes the associated allele to also be drawn. This should cause estimated coalescence times to be inflated, and estimated parameters should therefore be viewed as maximum estimates of the historical events. The inflated coalescence times are clearly seen in the estimated effective population sizes in S. troendelagicum populations seen presently (c. 110 000 individuals, compared with the census sizes of a few thousand individuals observed in natural populations). Therefore, it is likely that S. troendelagicum originated somewhat earlier than the modal value of the posterior estimates suggests. As the maximum age based on IM analyses and the minimum age of the ABC analyses are somewhat similar, we suggest that the species originated probably before the start-up of the Late Weichselian glaciation in Scandinavia (26 000 years bp) and no later than 80 000 years ago.
Conclusion
The parental species S. tenellum has a suboceanic distribution, whereas S. balticum occupies more continental areas, but they do coexist at many peatland localities in boreal areas of the Northern Hemisphere, particularly in Scandinavia. Both species are dioicous, but are commonly found with antheridia and sporophytes, even when they are found growing close together in the field. It is therefore surprising that so few populations of S. troendelagicum exist. Såstad et al. (2001) speculated that the difference in timing of gamete production in S. balticum and S. tenellum throughout most of their geographic distribution ranges could prevent hybridization events to occur and thus explain the limited distribution of S. troendelagicum. For hybridization to take place, the parental individuals must grow close together (Cronberg, 1996), but it is also possible that the spermatozoids can travel over longer distances with snow melting or rainwater, especially in sloped terrain. In any case, more work is needed to understand the unique occurrence of S. troendelagicum in central Norway.
The present results suggest that S. troendelagicum originated before the start of the Late Weichselian glacial period c. 26 000 years bp, implying that it must have had a different distribution previously than it does today. It could have survived the Late Weichselian glacial period in more southern or south-western parts of Europe not covered by ice. In any case, S. troendelagicum must have immigrated to central Norway quite recently, and then probably experienced a reduction in distribution area for some unknown reason, but most likely not because of its short evolutionary age. We have little reason to assume that this species will originate again in the future in the area that it is found today. To the contrary, habitat fragmentation and human pressure may cause the species to go extinct in foreseeable future. On the basis of our results, we suggest that all populations of the species be prioritized in future conservation programmes.
References
Abbott RJ, Lowe AJ (2004). Origins, establishment and evolution of new polyploidy species: Senecio cambrensis and S. eboracensis in the British Isles. Biol J Linn Soc 82: 467–474.
Adams KL (2007). Evolution of duplicate gene expression in polyploid and hybrid plants. J Hered 98: 136–141.
Beaumont MA (2008). Joint determination of topology, divergence time, and immigration in population trees. In: Matsura S, Forster P, Renfrew C (eds). Simulations, Genetics and Human Prehistory. McDonald Institute for Archaeological Research: Cambridge, pp 135–154.
Beaumont MA, Zhang W, Balding DJ (2002). Approximate Bayesian computation in population genetics. Genetics 162: 2025–2035.
Boatman DJ, Lark PM (1971). Inorganic nutrition of the protonemata of Sphagnum papillosum Lindb., S. magellanicum Brid. & S. cuspidatum Ehrh. New Phytol 70: 1053–1059.
Castric V, Bechsgaard J, Schierup M, Vekemans X (2008). Repeated adaptive introgression at a gene under multiallelic balancing selection. PLoS Genet 4: e1000168.
Chen ZJ (2007). Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol 58: 377–406.
Cornuet J-M, Santos F, Beaumont MA, Robert CP, Marin J-M, Balding DJ et al. (2008). Inferring population history with DIY ABC: a user-friendly approach to approximate Bayesian computation. Bioinformatics 24: 2713–2719.
Cronberg N (1996). Isozyme evidence of relationships within Sphagnum sect. Acutifolia (Sphagnaceae, Bryophyta). Plant Syst Evol 203: 41–64.
Daniels RE, Eddy A (1985). Handbook of European Sphagna. Institute of Terrestrial Ecology: Huntington.
Excoffier L, Laval G, Scheider S (2005). Arlequin ver 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Excoffier L, Smouse P, Quattro J (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Evanno G, Regnaut S, Gaudet J (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Fagundes NJR, Ray N, Beaumont MA, Neuenschwander S, Salzano F, Nonatto SL et al. (2007). Statistical evaluation of alternative models of human evolution. Proc Natl Acad Sci USA 104: 17614–17619.
Flatberg KI (1988). Sphagnum troendelagicum sp. nov. (sect. Cuspidata). Lindbergia 14: 22–39.
Flatberg KI, Blom HH, Hassel K, Kland RH (2006a). Moser Anthocerophyta, Marchatiophyta, Bryophyta. In: Kålås JA, Viken Å, Bakken T (eds). Norwegian Red list, Artsdatabanken: Trondheim. pp 416.
Flatberg KI, Thingsgaard K, Såstad SM (2006b). Interploidal gene flow and introgression in bryophytes: Sphagnum girgensohnii × S.russowii, a case of spontaneous neotriploidy. J Bryol 28: 27–37.
Fritsch R (1991). Index to bryophyte chromosome counts. Bryophytorum Bibliotheca 40: 1–352.
Gaeta RT, Chris Pires J (2009). Homoeologous recombination in allopolyploids: the polyploid ratchet. New Phytol 186: 18–28.
Garza JC, Williamson E. (2001). Detecting the reduction in population size using data from microsatellite DNA. Mol Ecol 10: 305–318.
Hey J, Nielsen R (2004). Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167: 747–760.
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009). Inferring weak population structure with the assistance of sample group information. Mol Ecol Res 9: 1322–1332.
Librado P, Rozas J (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Longton RE (1997). Reproductive biology and life-history strategies. In: Longton RE (ed). Advances in Bryology, Population Studies ol. 6. Gebrüder Borntraeger Verlagsbuchhandlung: Berlin and Stuttgart, pp 65–101.
Lundemo S, Falahati-Anbaran M, Stenøien HK (2009). Seed banks cause elevated generation times and effective population sizes of Arabidopsis thaliana in northern Europe. Mol Ecol 18: 2798–2811.
Mallet J (2007). Hybrid speciation. Nature 466: 279–283.
Martin NH, Bouck AC, Arnold ML (2006). Detecting adaptive trait introgression between Iris fulva and I. brevicaulis in highly selective field conditions. Genetics 172: 2481–2489.
McConnell R, Middlemist S, Scala C, Strassmann JE, Queller DC (2007). An unusually low microsatellite mutation rate in Dictyostelium discoideum, an organism with unusually abundant microsatellites. Genetics 177: 1499–1507.
Michalakis Y, Excoffier L (1996). A generic estimation of population subdivision using distances between alleles with special reference to microsatellite loci. Genetics 142: 1061–1064.
Miller N, Estoup A, Toepfer S, Bourguet D, Lapchin L, Derridj S et al. (2005). Multiple transatlantic introductions of the western corn rootworm. Science 310: 992.
Natcheva R, Cronberg N (2007). Maternal transmission of cytoplasmic DNA in interspecific hybrids of peat mosses, Sphagnum (Bryophyta). J Evol Biol 20: 1613–1616.
Nei M (1987). Molecular Evolutionary Genetics. Columbia University Press: New York.
Nielsen R, Wakeley J (2001). Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics 158: 885–896.
Ohta T, Kimura M (1973). A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genet Res 22: 201–204.
Pacak A, Szweykowaska-Kulinska Z (2000). Molecular data concerning alloploid character and the origin of the chloroplast and mitochondrial genomes in the liverwort species Pellia borealis. J Plant Biotechnol 2: 101–108.
Pascual M, Chapuis MP, Mestres F, Balanyà J, Huey RB, Gilchrist GW et al. (2007). Introduction history of Drosophila subobscura in the New World: a microsatellite-based survey using ABC methods. Mol Ecol 16: 3069–3083.
Pérez T, Albornoz J, Domínguez A (1998). An evaluation of RAPD fragment reproducibility and nature. Mol Ecol 7: 1347–1357.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rabouam C, Comes AM, Bretagnolle VV, Humbert JF, Periquet G, Bigot Y (1999). Features of DNA fragments obtained by random amplified polymorphic DNA (RAPD) assays. Mol Ecol 8: 493–503.
Ramos JR, Telles MP, Diniz-Filho JA, Soares TN, Melo DB, Oliveira G (2008). Optimizing reproducibility evaluation for random amplified polymorphic DNA markers. Genet Mol Res 7: 1384–1391.
Rannala B, Mountain JL (1997). Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94: 9197–9201.
Rieseberg LH (1997). Hybrid origins of plant species. Annu Rev Ecol Syst 28: 359–389.
Rieseberg LH, Willis JH (2007). Plant speciation. Science 317: 910–914.
Shaw AJ (2009). Bryophyte species and speciation. In: Goffinet B, Shaw AJ (eds). Bryophyte Biology. Cambridge University Press: Cambridge.
Shaw AJ, Andrus RE, Shaw B (2008a). Sphagnum beringiense sp. nov. (Bryophyta) from arctic Alaska, based on morphological and molecular data. Syst Bot 33: 469–477.
Shaw AJ, Cao T, Wang L-S, Flatberg KI, Flatberg B, Shaw B et al. (2008b). Genetic variation in three Chinese peat mosses (Sphagnum) based on microsatellite markers, with primer information and analysis of ascertainment bias. Bryologist 111: 271–281.
Shaw AJ, Cox CJ, Boles S (2003). Polarity of peatmoss (Sphagnum) evolution: who says mosses have no roots? Am J Bot 90: 1777–1787.
Slatkin M (1994). Linkage disequilibrium in growing and stable populations. Genetics 137: 331–336.
Solem T (1986). Age, origin and development of blanket mires in Sør-Trøndelag, Central Norway. Boreas 15: 101–115.
Soltis DE, Soltis PS (1993). Molecular data faciliate a reevaluation of traditional tenets of polyploid evolution. Crit Rev Plant Sci 12: 243–273.
Soltis DE, Soltis PS (1999). Polyploidy: recurrent formation and genome evolution. Trends Ecol Evol 14: 348–352.
Soltis DE, Soltis PS, Pires JC, Kovarik A, Tate J, Mavrodiev E (2004). Recent and recurrent polyploidy in Tragopogon (Asteraceae): cytogenetic, genomic and genetic comparisons. Biol J Linn Soc 82: 485–501.
Soltis DE, Soltis PS, Tate JA (2003). Advances in the study of polyploidy since Plant speciation. New Phytol 161: 173–191.
Soltis PS, Soltis DE (2009). The role of hybridization in plant speciation. Annu Rev Plant Biol 60: 561–588.
Stenøien HK, Fenster CB, Tonteri A, Savolainen O (2005). Genetic variability in natural populations of Arabidopsis thaliana in northern Europe. Mol Ecol 14: 137–148.
Stenøien HK, Flatberg KI (2000). Genetic variability in the rare Norwegian peat moss Sphagnum troendelagicum. Bryologist 103: 794–801.
Stenøien HK, Såstad SM (1999). Genetic structure in three haploid peat mosses (Sphagnum). Heredity 82: 391–400.
Stenøien HK, Såstad SM (2001). Genetic variability in bryophytes: does mating system really matter? J Bryol 23: 313–319.
Sundberg S (2005). Larger capsules enhance short-range spore dispersal in Sphagnum, but what happens further away? Oikos 108: 115–124.
Szövényi P, Terracciano S, Ricca M, Shaw AJ (2008). Recent divergence, intercontinental dispersal and shared polymorphisms are shaping the genetic structure of amphi-Atlantic peatmoss populations. Mol Ecol 17: 5364–5377.
Såstad SM (2004). Patterns and mechanisms of polyploid speciation in bryophytes. Regnum Veget 142: 317–333.
Såstad SM, Stenøien HK, Flatberg KI, Bakken S (2001). The narrow endemic Sphagnum troendelagicum is an allopolyploid derivative of the widespread S. balticum and S. tenellum. Syst Bot 26: 66–74.
Udupa SM, Baum M (2001). High mutation rate and mutational bias at (TAA) (n) microsatellite loci in chickpea (Cicer arietinum L.). Mol Genet Genomics 265: 1097–1103.
Van Zanten BO (1978). Experimental studies on trans-oceanic long-range dispersal of moss spores in the Southern Hemisphere. J Hattori Bot Lab 44: 455–582.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Whithney KD, Randell RA, Rieseberg LH (2006). Adaptive introgression of herbivore resistance traits in the weedy sunflower Helianthus annuus. Am Nat 167: 794–807.
Williams JG, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18: 6531–6535.
Wolfe KH, Li W-H, Sharp PM (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci USA 84: 9054–9058.
Wood TE, Takebayashi N, Barker MS, Mayrose I, Greenspoon PB, Rieseberg LH (2009). The frequency of polyploid speciation in vascular plants. Proc Natl Acad Sci USA 106: 13875–13879.
Acknowledgements
We thank three reviewers and the editor for comments and suggestions that improved the paper. This research was supported by NSF Grant DEB-0918998 to AJS and BS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Stenøien, H., Shaw, A., Stengrundet, K. et al. The narrow endemic Norwegian peat moss Sphagnum troendelagicum originated before the last glacial maximum. Heredity 106, 370–382 (2011). https://doi.org/10.1038/hdy.2010.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.96
