
Abstract
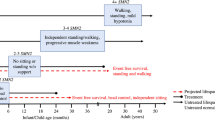
SMA is a rare hereditary neuromuscular disease that causes weakness and muscle wasting as a result of the loss of spinal motor neurons. In its most severe form, SMA is the commonest genetic cause of death in infants, and children with less severe forms of SMA face the prospect of lifelong disability from progressive muscle wasting, loss of mobility and limb weakness. The initial discovery of the defective gene has been followed by major advances in our understanding of the genetic, cellular and molecular basis of SMA, providing the foundation for a range of approaches to treatment, including gene therapy, antisense oligonucleotide treatments and more traditional drug-based approaches to slow or halt disease progression. The approval by the US Food and Drug Administration (FDA) of Spinraza (nusinersen), the first targeted treatment for spinal muscular atrophy (SMA), is a historic moment. Disease-focused research charities, such as The SMA Trust (UK), continue to have a crucial role in promoting the development of additional treatments for SMA, both by funding translational research and by promoting links between researchers, people living with SMA and other stakeholders, including pharmaceutical companies and healthcare providers.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

References
Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80: 155–165.
Burghes AH, Beattie CE . Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 2009; 10: 597–609.
Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999; 8: 1177–1183.
Feldkotter M, Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B . Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002; 70: 358–368.
Riessland M, Kaczmarek A, Schneider S, Swoboda KJ, Löhr H, Bradler C et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet 2017; 100: 297–315.
Shababi M, Glascock J, Lorson CL . Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum Gene Ther 2011; 22: 135–144.
Hosseinibarkooie S, Peters M, Torres-Benito L, Rastetter RH, Hupperich K, Hoffmann A et al. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am J Hum Genet 2016; 99: 647–665.
Hammond SM, Hazell G, Shabanpoor F, Saleh AF, Bowerman M, Sleigh JN et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci USA 2016; 113: 10962–10967.
Dominguez E, Marais T, Chatauret N, Benkhelifa-Ziyyat S, Duque S, Ravassard P et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet 2011; 20: 681–693.
Benkhelifa-Ziyyat S, Besse A, Roda M, Duque S, Astord S, Carcenac R et al. Intramuscular scAAV9-SMN injection mediates widespread gene delivery to the spinal cord and decreases disease severity in SMA mice. Mol Ther 2013; 21: 282–290.
Tanguy Y, Biferi MG, Besse A, Astord S, Cohen-Tannoudji M, Marais T et al. Systemic AAVrh10 provides higher transgene expression than AAV9 in the brain and the spinal cord of neonatal mice. Front Mol Neurosci 2015; 8: 36.
Bucher T, Dubreil L, Colle MA, Maquigneau M, Deniaud J, Ledevin M et al. Intracisternal delivery of AAV9 results in oligodendrocyte and motor neuron transduction in the whole central nervous system of cats. Gene Ther 2014; 21: 522–528.
Duque SI, Arnold WD, Odermatt P, Li X, Porensky PN, Schmelzer L et al. A large animal model of spinal muscular atrophy and correction of phenotype. Ann Neurol 2015; 77: 399–414.
Valori CF, Ning K, Wyles M, Mead RJ, Grierson AJ, Shaw PJ et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med 2010; 2: 35ra42.
Lutz CM, Kariya S, Patruni S, Osborne MA, Liu D, Henderson CE et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Invest 2011; 121: 3029–3041.
Kariya S, Obis T, Garone C, Akay T, Sera F, Iwata S et al. Requirement of enhanced survival motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest 2014; 124: 785–800.
Ning K, Drepper C, Valori CF, Ahsan M, Wyles M, Higginbottom A et al. PTEN depletion rescues axonal growth defect and improves survival in SMN-deficient motor neurons. Hum Mol Genet 2010; 19: 3159–3168.
Powis RA, Karyka E, Boyd P, Côme J, Jones RA, Zheng Y et al. Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 2016; 1: 1–16.
Wishart TM, Mutsaers CA, Riessland M, Reimer MM, Hunter G, Hannam ML et al. Dysregulation of ubiquitin homeostasis and beta-catenin signaling promote spinal muscular atrophy. J Clin Invest 2014; 124: 1821–1834.
Sleigh JN, Gillingwater TH, Talbot K . The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy. Dis Model Mech 2011; 4: 457–467.
Hamilton G, Gillingwater TH . Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med 2013; 19: 40–50.
Wirth B, Barkats M, Martinat C, Sendtner M, Gillingwater TH . Moving towards treatments for spinal muscular atrophy: hopes and limits. Expert Opin Emerg Drugs 2015; 20: 353–356.
Thomson AK, Somers E, Powis RA, Shorrock HK, Murphy K, Swoboda KJ et al. Survival of motor neurone protein is required for normal postnatal development of the spleen. J Anat 2017; 230: 337–346.
Szunyogova E, Zhou H, Maxwell GK, Powis RA, Francesco M, Gillingwater TH et al. Survival Motor Neuron (SMN) protein is required for normal mouse liver development. Sci Rep 2016; 6: 34635.
Sintusek P, Catapano F, Angkathunkayul N, Marrosu E, Parson SH, Morgan JE et al. Histopathological defects in intestine in severe spinal muscular atrophy mice are improved by systemic antisense oligonucleotide treatment. PLoS ONE 2016; 11: e0155032.
Somers E, Lees RD, Hoban K, Sleigh JN, Zhou H, Muntoni F et al. Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann Neurol 2016; 79: 217–230.
Sleigh JN, Barreiro-Iglesias A, Oliver PL, Biba A, Becker T, Davies KE et al. Chondrolectin affects cell survival and neuronal outgrowth in in vitro and in vivo models of spinal muscular atrophy. Hum Mol Genet 2014; 23: 855–869.
Little D, Valori CF, Mutsaers CA, Bennett EJ, Wyles M, Sharrack B et al. PTEN depletion decreases disease severity and modestly prolongs survival in a mouse model of spinal muscular atrophy. Mol Ther 2015; 23: 270–277.
Acknowledgements
We thank Dr Lynn Ossher for reviewing this paper as well as all our supporters for all their donations which make our work possible.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Christie-Brown, V., Mitchell, J. & Talbot, K. The SMA Trust: the role of a disease-focused research charity in developing treatments for SMA. Gene Ther 24, 544–546 (2017). https://doi.org/10.1038/gt.2017.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2017.47
