
Abstract
Purpose:
Cell-free DNA (cfDNA) testing for fetal aneuploidies was broadly implemented for common trisomies and sex-chromosome anomalies (SCAs). However, such an approach identifies only 75 to 85% of clinically relevant aneuploidies.
Methods:
We present a consecutive series of 6,388 cases, thus uncovering a broader array of aneuploidies, including the rare autosomal trisomies (RATs) and the maternally inherited deletion and duplication copy-number variations (CNVs), with complete and stratified follow-up by amniocentesis. Combined measurements of z-scores and the fetal fraction, in conjunction with fetal cfDNA enrichment, were used to stratify the likelihood of true and false results.
Results:
We obtained an incremental diagnostic yield of 50%; RATs and CNVs were found to be significant causes of fetal pathology. Scrutinizing z-scores and the fetal fraction made it possible to distinguish the sources of false-negative results; predict the likelihood of false-positive results for major trisomies and SCAs; classify maternal mosaic SCAs and CNVs, preventing false-positive results; and robustly identify maternally inherited CNVs and detect recurrent genomic disorders as a standardized function of the fetal fraction.
Conclusion:
With the clinical pertinence of this broader detection scheme confirmed, we offer recommendations for its implementation.
Genet Med 19 2, 169–175.
Similar content being viewed by others

Introduction
Large-scale clinical studies and an updated meta-analysis comprehensively described the performance of cfDNA testing for the common trisomies—21, 18, and 13—and, to a lesser extent, for sex-chromosome anomalies (SCAs).1,2,3,4 Independent lines of converging evidence suggest that a substantial fraction of clinically relevant aneuploidies is neglected by such a restricted detection scheme: (i) the common trisomies comprise only approximately 75% of aneuploidies detected by karyotyping in Down syndrome screen-positive cases, with 85% including the SCAs5; (ii) registry data showed that 17% of clinically relevant anomalies would go undetected6; and (iii) a review of 4,000 prenatal karyotype results revealed that 24% of the reported anomalies would have been overlooked.7
The main aneuploidy classes ignored by the current generation of cfDNA screening methods comprise the rare autosomal trisomies (RATs) and the structural chromosome anomalies, specifically disease-causing copy-number variations (CNVs). Both types of aberrations generate positive results in conventional aneuploidy screening used to prescribe cfDNA testing. In fact, first-trimester screening for T21, T18, and T13 is also sensitive to a broad range of RATs and chromosomal mosaicism.8 Chromosomal microarray platforms provide an incremental diagnostic yield of 5% in fetuses with increased nuchal translucency (NT) diagnosed by first-trimester ultrasound and a normal karyotype.9
Independent follow-up of pregnancies considered unaffected by such cfDNA tests confirmed that rare pathogenic aneuploidies such as T22 mosaicism are overlooked.10 Given the significant incidence of mosaic aneuploidy,11,12 and the fact that the knowledge base on rare trisomy mosaicism has considerably increased,13 routine detection of such anomalies in mainstream cfDNA clinical practice becomes an option. Moreover, the phenotypic impact of RATs reaches beyond fetal mosaic trisomies; after trisomy or monosomy rescue, uniparental disomy (UPD) can cause fetal pathology, even in diploid fetuses when chromosomes undergoing imprinting are involved.12,14 Placental trisomies account for a low but significant risk of intrauterine growth restriction, and for an even higher risk of small-for-gestational-age infants.15
The population history16 and morbidity maps17 of deletion and duplication CNVs have been extensively described. It has been estimated that CNVs >400 kb account for almost 15% of disease burden in children affected by intellectual disability and congenital anomalies.17 A meta-analysis confirmed the incremental diagnostic yield of performing chromosomal microarray analysis for cases of increased NT after first-trimester ultrasound.9 Implementation of routine CNV detection using cfDNA sequencing posed challenges, however. Initial studies suggested that higher genomic coverage was required to detect CNVs.18,19 Also, experimental protocols that robustly detect defined CNV size classes that take the fetal fraction into account and use genomic coverage similar to that of numerical anomalies were not available until recently.20 A clinical follow-up study effectively showed that cfDNA testing can be extended to include reliable detection of deletion CNVs.21
We previously described a technical validation study using cfDNA screening with low genomic coverage that robustly detected a broader array of anomalies comprising the common trisomies, the SCAs, the RATs, and deletion and duplication CNVs.22 The algorithms were optimized for uniformly reliable detection of numerical autosomal and sex-chromosome anomalies and CNVs, in a manner similar to procedures described in a recent report.23
Here, we review a series of 6,388 consecutive singleton pregnancies with nearly complete and stratified follow-up; we tested the extended range of anomalies. Moreover, fetal fraction measurements and a fetal cfDNA enrichment procedure were integrated. The latter was used in conjunction with signal intensity/z-scores to determine the main sources of false-negative results (low fetal fraction and true fetal mosaicism/TFM5), to predict maternally inherited numerical SCAs and CNVs, to stratify the likelihood of true-positive versus false-positive results, and to establish the detection threshold for recurrent pathogenic CNVs. We issue recommendations for clinical implementation of such a detection scheme on a broader scale.
Materials and Methods
Study design
The consecutive cases consisted of two nonoverlapping data sets. The first aimed at complete follow-up of newborns based on the predicted birth of all pregnancies included, and the second aimed at evaluating the effects of integrating the routine measurement of the fetal fraction. The fetal fraction (methodology described in the supplementary section) was retroactively assessed for the first set. All surviving pregnancies of the second set were born. The principal outcomes were the accurate classification of singleton pregnancies as euploid and aneuploid and the follow-up of pregnancies with invasive testing/amniocentesis for aneuploidies considered aberrant/abnormal, likely aberrant/abnormal, or of unknown clinical significance (UNK). Clinical outcomes were ascertained at birth for euploid results considered normal, likely normal, and UNK. The follow-up was stratified. The proportion of cases followed-up with amniocentesis performed by internal or external cytogenetics laboratories increased with increasing uncertainty about the abnormality status. Pregnancy outcomes were monitored by using online registries for verified aneuploidies and for birth outcomes based on voluntary information provided by physicians, by inquiring about the birth outcomes of two sets of 250 random samples in the two main linguistic regions, and by consulting with other physicians. The classification of test results is described in the Supplementary Materials and Methods online.
Samples, cfDNA aneuploidy screening, fetal cfDNA enrichment, data sets, and patients
Ethylenediaminetetraacetic acid blood tubes (Becton Dickinson, Sarstedt, Germany) were used because of regulatory requirements (CE marking). Exclusion criteria were as follows: transportation time >48 h, total DNA concentrations ≥4 ng/μl, and visible hemolysis (degree defined by photographic references). The cfDNA extracted from 1 ml of plasma was analyzed by shotgun sequencing on Illumina sequencers (HighSeq 2000),22 with a minimal genomic coverage of 0.0156-fold (≥10 × 106 unique exact 50-nt sequence tags). Fetal cfDNA enrichment was obtained by removing fragments >200 nt by gel electrophoresis (methodology described in supplementary section). Two consecutive data sets based on test reports by board-certified laboratory geneticists were retrieved from the clinical database: one for the period beginning in March 2013 through August 2014 and one from the beginning of September 2014 through May 2015, which was after the integration of the routine measurement of the fetal fraction.
The first data set included 4,545 pregnancies (4,497 singleton and 48 twin pregnancies) the second included 1,843 samples (not overlapping with the first set). The results for annotated singleton pregnancies were used as the basis for the statistics after one additional review by an independent expert geneticist. Samples with fetal fractions <3% were excluded and a second blood draw was requested.
Results
Readout and follow-up
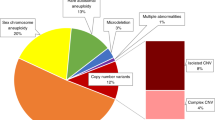
Overall, 258/6,388 (4.04%) samples were considered abnormal or likely abnormal ( Figure 1 ), comprising 119 common trisomies (1.86%), 53 SCAs (0.83%), 50 RATs (0.78%), and 36 CNVs (0.56%). Conversely, 6,130 samples (95.96%) were considered normal or likely normal. Details on the sample and patient characteristics can be found in the Supplementary Results online section.

The detected aneuploidy classes. The absolute numbers of the aneuploidy classes are shown from left to right: the common trisomies (trisomy 21, 18, and 13), the sex-chromosome anomalies (monosomy X, triple X, and Klinefelter syndromes), the rare autosomal trisomies, and the deletion and duplication copy-number variations.
The 119 common trisomies were invariably classified as abnormal or likely abnormal and comprised 84 T21 (70.6%), 19 T18 (T18; 16%), and 16 T13 samples (13.4%). Fifteen additional samples were rated as UNK (0.23%). Discriminating between euploid and aneuploid was simple for T21 because 75/84 (89.3%) z-scores were at least 1.5 times higher than the threshold. All but three results (81/84; 97.6%) were considered aberrant—these three were rated likely aberrant—and three were classified as UNK. Trisomy 18 scores were also generally high (≥1.5× threshold z-score) for 16/19 (84.2%), with 18/19 rated abnormal and 1 rated likely abnormal, but a higher number, 8 overall, were considered UNK. Twelve out of 16 T13 results (75%) had high scores (≥1.5× threshold z-score), 15/16 were aberrant, 1 was likely aberrant, and 4 were considered UNK.
Follow-up with amniocentesis was 47% for the aberrant/likely aberrant group (57/121) and 80% for the UNK group (12/15). The workup of the newborn babies who tested “normal/likely normal” did not yield discrepant results. The UNK samples without follow-up all belonged to the first data set and were from newborns who were born healthy (composite follow-up of UNK samples 15/15 or 100%). For T21, there were the following discrepancies (henceforth designated as “FPs” or “FNs”): four false positives (FPs) and no false negatives (FNs); two FNs and no FP for T18; and four FPs and no FN for trisomy 13 ( Table 1 ). Thus, the estimates for the false-positive rates (FPRs; Table 1 ) were 0.063% for T21, <0.001% for T18, and 0.062% for T13. The detection rates (DRs; Table 1 ) were >99.99% for T21 and T13 (84/84 and 16/16, respectively) and approximately 90% for T18 (17/19). Three out of three with UNK status for chromosome 21 and four out of four with UNK status for chromosome 13 turned out to be normal diploids. Six out of eight with UNK status for chromosome 18 turned out to be normal diploids, of which two babies were born healthy without aneuploidy and two had false-negative results.
The reasons for the two FNs for T18 were low fetal fraction in one case and likely TFM5 for the other case. Both samples were in the first data set, before introduction of routine fetal fraction measurements. The first FN with a fetal fraction of 3.7% and native z-score of 3.1 showed complete and specific rescue of an abnormal T18 signal after cfDNA enrichment to 33.4% with a z-score of 7.2; the T18 signal of the second FN with a native fetal fraction of 9.5% and z-score of 2.9, however, stayed close to, but did not cross the threshold even after enrichment to 28.5% and a z-score 2.7. A similar enrichment of euploid samples did not generate FP T18 results. The first FN was therefore caused by low fetal fraction. For the second, there was only a limited amount of cfDNA molecules with T18 available in the circulating blood, indicating TFM5.
Low z-scores in the presence of average fetal fractions were the best predictor of FPs. Overall there were eight FPs: four for T21, four for T13, and none for T18. The key difference between true positives (TPs) and FPs is that TPs (n = 10) had significantly higher z-scores than the FPs (n = 7) yet similar fetal fractions (z-scores and fetal fractions for TPs/FPs were 15.6 ± 3.7/4.97 ± 0.79 (t-test P < 0.0001) and 11.6 ± 1.91%/10.2 ± 1.75% (P = 0.1623)). One FP for T21 was indistinguishable from TPs based solely on the z-score of 29.8 and the fetal fraction of 25%.
If low-grade placental confined mosaicism (CPM) is the principal reason for FPs, then the trisomy z-scores will increase only marginally, even for maximal fetal fractions of 50%, and this is exactly what was found. For five FPs, the fetal fraction was enriched from 10.28 ± 1.56% to 51.08 ± 8.2% (P < 0.0001) and the corresponding z-scores moved from 5.11 ± 0.93 to 7.74 ± 2.97 (P = 0.0964). For one single FP with an initial z-score of 29.8, the enrichment from 25% to 56.7% moved the z-score to 54.25. This indicates that low z-scores as a proxy for low-grade CPMs are a more frequent cause of FPs than high z-scores as proxy for high-level mosaicism, provided that the fetal fractions are comparable and average for both, in accordance with predictions based on CVS data.12,24,25,26
For two FPs, however, the z-score was not increased (0.7 and 1.58) despite significant augmentation of the fetal fraction from 12.6 to 60.8% and from 11.1 to 54.6%. For the latter two, a technical problem (i.e., signal variance) is the most likely explanation.
Fifty-three cases (0.83%; 53/6,388) of SCAs were found ( Figure 1 ): 38 (71%) with monosomy X (MX), 10 (18.9%) with triple X syndrome (TXS), and 5 (9.4%) with Klinefelter syndrome (KS). Inversely, 99.17% of samples had a physiological sex-chromosome dosage. Of the 38 MX cases, 32 were classified as abnormal and 6 were classified as likely abnormal. Four out of those 32 considered abnormal had z-scores largely superior (P < 0.001) to those obtained for the highest fetal fractions, suggesting maternal MX mosaicism. The four candidate maternal MX cases had fetal fractions and z-scores of 8.8% ± 4.25 and −30.7 ± 11.11. Compared to the confirmed exclusively fetal reference MX cases (n = 15; z-scores −9.28 ± 3.56), the values for the candidate maternal MX mosaicism cases are expected to occur for less than one in a million exclusively fetal cases with 45,X (z-scores <−26.7). All 10 TXS and all 5 KS were considered abnormal. One TXS had a z-score largely exceeding those of the highest fetal fractions, indicating maternal TXS. The z-scores for the validated fetal reference 47,XXX cases were 12.1 ± 2.29 (n = 5) and the candidate maternal 47,XXX case had a z-score of 75.3; such values are expected for less than one in a trillion cases with purely fetal 47,XXX (z-scores >28.4).
Follow-up with amniocentesis was 63% (24/38) for MX, 40% (4/10) for KS, and 20% (1/5) for TXS. The inquiry of suspected newborns did not reveal clinically evident cases present at birth or the presence of classic Turner syndrome. Specifically, three potential MX cases were followed up at birth, two FPs with normal postnatal karyotype 46,XX and one UNK who was clinically unsuspicious (total follow-up: 27/39, 69%). There were no known FNs and 17 FPs for MX, and no known FNs and FPs for TXS and KS (Supplementary Table S1 online).
Low z-scores concomitant with average fetal fractions were—similar to the common trisomy group—a reliable predictor of FPs. This is supported by the following lines of evidence: (i) the likelihood of confirming a positive 45,X test result with amniocentesis increases with the z-score as a proxy for the degree of mosaicism—the z-scores and fetal fractions of TPs (n = 5)/FPs (n = 6) were −10.12 ± 1.43/−5.28 ± 1.47 (P = 0.0004) and 15.8 ± 9.7%/13.9 ± 2.9%; (ii) specifically and strongly increased z-scores after a second blood draw or experimental fetal cfDNA enrichment are predictive of TPs after amniocentesis (z-scores and fetal fractions for first/second blood draw were −4.26/−9.36 and 5.6%/15.3%, and before/after fetal cfDNA enrichment were −6.14/−12.4 and 8.6%/29.7%, respectively); (iii) 15 out of 15 clinically affected 45,X cases were all correctly called in a blinded fashion during our technical validation study.22 Globally, the likelihood of positive cfDNA 45,X test calls to be confirmed by amniocentesis is largely dominated by biological parameters, of which CPM is the single most important one, as predicted by CVS data.12,24,25,26
The higher prevalence of FPs for MX compared to the other SCA and common trisomies is expected because the CPM1/3 rate is much higher, approximately 60% for MX and mosaicism levels exceeding 50%.26 This fully accounts for the positive predictive value (PPV) of 55% observed for MX (Supplementary Table S1 online). The FPR for MX is 0.26%, the FPRs for the other SCAs for both KS and TXS are <0.01%. The DRs for the SCAs were nominally >99.9%, specifically 21/21 for MX (9/9 directly validated), 10/10 for KS, and 5/5 for TXS. This excludes only mosaic aneuploidies that are clinically detectable at birth.
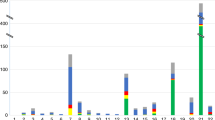
In the RAT group, 50 cases were found (50/6,388, 0.78%; Figure 2 ). These are listed individually in Figure 2 : the most frequent (32%) was T7, with 16 cases that could be associated with UPD.14 The group with a high or very high risk for an unfavorable outcome if the fetus were affected13 comprised seven cases (14%). Trisomy cases potentially associated with UPD, namely T6, T7, T14, T15, and T16, were considered abnormal or likely abnormal because UPD can be symptomatic even in diploid fetuses after trisomy or monosomy rescue.14 All other trisomies were rated abnormal or likely abnormal based on the relative evidence for further workup.6,13 Overall, 35 out of 50 (70%) trisomies were considered abnormal, and 15 (30%) were considered likely abnormal.

The detected individual rare autosomal trisomies. The absolute numbers of the individual rare autosomal trisomies are shown from left to right in decreasing order of frequency found.
The follow-up with amniocentesis was 38% for the group as a whole (19/50) and focused on critical groups, for instance: 100% (3/3) for trisomy 22, 50% (2/4) for trisomy 16, and 37.5% (6/16) for T7, which included routine molecular UPD analysis in addition to karyotyping. Four fetal aneuploidies were confirmed; all three T22 mosaicism cases were fetal, as was one case of T12 mosaicism. For all remaining cases, amniocentesis revealed normal diploid results; in the cases with potential UPD, no single fetal UPD was identified. This resulted in a nominal FPR of 0.71% and a low PPV of 8%.
Several lines of evidence support the notion that these low PPVs reflect primarily CPM24 rather than technical problems. First, the z-scores between the TPs and FPs did not significantly differ. We compared the TPs, three with T22 and one with T12 mosaicism, with the FPs comprising the trisomies having the highest risk for an unfavorable outcome,13 namely T7, T9, T16, and T20. The z-scores and fetal fractions of TPs/FPs were not significantly different: 12 ± 3.8/10.72 ± 6.42 (P = 0.73) and 11.05 ± 2.16%/11.17 ± 4.25% (P = 0.95). Second, stronger increases in z-scores on cfDNA enrichment than those observed for the FPs of the common trisomies were observed (z-score and fetal fraction native/enriched were 5.18/11.91 and 5.8%/55.3%). For predicted cases of T16 mosaicism, a consistent rate of unfavorable pregnancy outcome was observed (25%) as expected.15 The technical validation study had proven highly accurate detection of this class of anomalies.22
Thirty-six candidate CNVs were identified, 25 in the first data set and 11 in the second data set (0.56%) ( Table 2 ). All were rated pathogenic or likely pathogenic without clear-cut distinction because of the difficulty of integrating multiparametric factors such as the critical regions, size, penetrance, and inheritance. Copy-number variation in two genomic regions known to undergo recurrent rearrangements and to overlap with well-known genomic disorders were more frequently found, namely, two deletions and two duplications of the DGS region 22q11.2 (11%) and two deletions and two duplications of the 16p13.11 critical region (11%) ( Table 2 ).17 Other recurrent CNVs were identified, such as two duplications of the 15q11.2 region and one duplication of the 17p12 region ( Table 2 ). Nonrecurrent CNVs not estimated to overlap with known genomic disorders are listed in Table 2 .
For 11/36 (30.5%), follow-up data with chromosomal microarray and/or FISH analysis after amniocentesis and parental analyses were available. For the recurrent CNVs with defined pathogenicity, the follow-up was nearly complete; specifically, it was available for 3/4 CNVs of the 22q11.2 region and for 3/4 CNVs of the 16p13.11, as well as for the duplication of 17p12 (PMP22). The following maternally transmitted CNVs were all confirmed in mother and fetus: two duplications and one deletion of 22q11.2, one duplication and one deletion of 16p13.11, and one duplication of 17p12. In the nonrecurrent CNV class, a maternally transmitted deletion of 9q31 was confirmed in mother and fetus. Overall, three candidate CNVs were not confirmed: one of the deletions of 16p13.11 and, in the nonrecurrent CNV class, a deletion of 5q14.3q23.2 and a deletion of 10q22-q33, totaling three FP results. During the follow-up of pregnancies in the first data set, prior to fetal fraction measurements, a 10-Mb terminal deletion of 4p had not been called (one FN result).
Based on this experience, the analytical DRs were defined experimentally for recurrent CNVs of defined size and as a function of the fetal fraction ( Figure 3 ). These experiments indicate a robust detection of CNVs with a size class of 3 Mb for the most frequently implicated region, 22q11.2, and an average range of fetal fractions of 10%. Accordingly, we recommend limiting the clinical use to detection of recurrent CNVs of defined size and penetrance17 for which the DR can be defined with the use of positive controls and depending on the fetal fraction.

Standardization of detection of Di George syndrome duplication. Average (±1 SD) signal intensities of the negative control (left) and of serial dilutions (from left to right: 5%, 10%, 15%, 20%, 25%) of the chromosomal microarray-validated fetal case of maternal origin with a 3-Mb duplication of 22q11.2 (shown as 100% dosage on the far right). The detection thresholds are shown as dashed lines, the lower line represents the 99% confidence level (z-score +2.575829), and the upper line reflects the 99.9% confidence level (z-score +3.290527).
Discussion
Here, we reviewed the follow-up of a series of 6,388 consecutive clinical cases using cfDNA screening with low genomic coverage and detected a broad range of aneuploidy classes, namely the common trisomies, the SCA, the RATs, and deletion and duplication CNVs.
Clinical follow-up amniocentesis results considered abnormal, likely abnormal, or UNK were between 30 and 80% for the four aneuploidy categories (mean, 42.5 ± 10.8%). It was generally close to complete—between 75 and 80%—for results considered UNK and when maternal mosaicism was suspected. Follow-up for results classified as normal or likely normal was ensured by an online birth registry, by an inquiry of randomly selected samples, and by proximity to the addressing physicians. These follow-up figures typically are higher than those reported in the literature, especially for the UNK group.
In comparison with the most recently updated meta-analysis,1 we report both very high DRs and very low FPRs for the common trisomies. Overall, only two FNs were recorded (both for T18): one caused by a low fetal fraction that would be avoided under current conditions and the other due to likely TFM5. A lower DR for T18 compared to T21 and T13 is exactly what is expected because of the higher TFM rate for T18 based on CVS data.26 The workup of the eight FPs for T21 and T13 showed that low z-scores as a proxy for likely low-grade CPMs are more frequently producing FPs than high z-scores as a proxy for high-level mosaicism, provided there are comparably normal fetal fractions. These findings, again, precisely reflect the predictions of CVS data that relate four times more FPs to low-grade than high-grade trisomy mosaicism.12,24-26 We did not find any evidence of maternal CNVs as potential confounding factors, as reported by a number of studies.2,23,27,28 The proportion of FPs explained by a demising twin is difficult to access because there is no systematic measurement of the number of heartbeats in early pregnancy. The key conclusions are, first, that the specific nature of FNs (low fetal fraction versus TFM5) and, second, the likelihood of FPs can be predicted based on z-scores, fetal fraction, and cfDNA enrichment.
Parallel evidence is provided for the accurate detection of SCAs, consistent with our technical validation study demonstrating performance similar to the common trisomies.1,22 In accordance with the predictions of CVS data;24,26 however, CPM was responsible for a significant proportion of FPs likely accounting for 37% of all MX results. Vanishing twins again constitute a mystifying cause that could not be safely dissociated from placental mosaicism. Interestingly, 3 out of the 17 FPs comprising 7.9% of all MXs were likely due to maternal MX mosaicism, based on two lines of evidence: (i) exceedingly high z-scores incompatible with those expected for purely fetal affections and (ii) although these three cases were not worked-up, a sample indistinguishable from the three based on the z-scores and fetal fraction was shown to correspond to 14% maternal MX mosaicism (mos 45,X[7]/46,XX[43]). In conclusion, detection of SCAs presents evidence similar to that of the common trisomies, provided that (i) an adequate technical validation study is corroborated by clinical follow-up programs; (ii) detection is limited to aneuploidies with evidence-based workup; and (iii) women are counseled appropriately because of higher CMP rates and significant risk of detecting maternal mosaicism.
Based on the current data series, we suggest that detection of RATs is clinically useful for at least five reasons: (i) the accuracy was similar to that of the common trisomies and SCAs22; (ii) the PPVs closely reflected those predicted by CVS data;13 (iii) T22 mosaicism is an important cause of truly fetal aneuploidy; (iv) although UPD cases were not detected in the present study, this reflects prevalence data;13 and (v) even without UPD, placental mosaicism carries a small but definite risk for intrauterine growth restriction, small-for-gestational-age infants, and unfavorable pregnancy outcome (e.g., T16), which is supported by this study.15
We initially designed CNV detection to be genome-wide in principle. The experience with these data set clearly revealed the limitations of such an approach; for most, if not all, nonrecurrent CNVs, solid and reliable data on allele frequencies, segregation, implicated genes and pathways, and phenotypic consequences are lacking, which makes clinical implementation in a prenatal setting problematic. For this reason, the diagnostic workup of recurrent CNVs was emphasized. This experience clearly showed that this subclass of CNVs can be robustly detected in the context of a routine clinical program. Specifically, spike-in experiments ( Figure 3 ) using positive control cfDNA allowed the detection sensitivity to be defined for CNV size classes dependent on the fetal fraction. Similar to what we described for the SCAs, the signal intensity could also be used to predict whether a CNV is maternally transmitted. Consequently, such predictions can be implemented to prevent false-positive CNV calling.27
The following recommendations can be made based on the present study: (i) cfDNA screening should be extended to include detection of RATs and deletion/duplication-CNVs; (ii) integrated interpretation of the fetal fraction, z-scores, and fetal cfDNA enrichment should be used to stratify the likelihood of FNs/FPs caused by CPM and maternal aneuploidy/CNVs; and (iii) CNV detection should be based on experimental validation of defined CNV size classes integrating the fetal fraction and should be limited to a list of well-characterized genomic disorders. The latter can be expanded with the increasing knowledge base. Extensions of the present study could comprise targeted high-coverage sequencing to more closely investigate UPD and the potential recessive unmasking associated with it, as well as to search for frequent single-gene disorders in the future.
Disclosure
G.P., M.O., L.F., and B.C. are minority shareholders of Sonic Healthcare, which owns Aurigen, Fasteris, and Genesupport. The other authors declare no conflict of interest.
References
Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: updated meta-analysis. Ultrasound Obstet Gynecol 2015;45:249–266.
Zhang H, Gao Y, Jiang F, et al. Non-invasive prenatal testing for trisomies 21, 18 and 13: clinical experience from 146,958 pregnancies. Ultrasound Obstet Gynecol 2015;45:530–538.
McCullough RM, Almasri EA, Guan X, et al. Non-invasive prenatal chromosomal aneuploidy testing–clinical experience: 100,000 clinical samples. PLoS One 2014;9:e109173.
Bianchi DW, Parsa S, Bhatt S, et al. Fetal sex chromosome testing by maternal plasma DNA sequencing: clinical laboratory experience and biology. Obstet Gynecol 2015;125:375–382.
Davis C, Cuckle H, Yaron Y. Screening for Down syndrome–incidental diagnosis of other aneuploidies. Prenat Diagn 2014;34:1044–1048.
Wellesley D, Dolk H, Boyd PA, et al. Rare chromosome abnormalities, prevalence and prenatal diagnosis rates from population-based congenital anomaly registers in Europe. Eur J Hum Genet 2012;20:521–526.
Lebo RV, Novak RW, Wolfe K, Michelson M, Robinson H, Mancuso MS. Discordant circulating fetal DNA and subsequent cytogenetics reveal false negative, placental mosaic, and fetal mosaic cfDNA genotypes. J Transl Med 2015;13:260.
Tørring N, Petersen OB, Becher N, Vogel I, Uldbjerg N ; Danish Fetal Medicine Study Group; Danish Clinical Genetics Study Group. First trimester screening for other trisomies than trisomy 21, 18, and 13. Prenat Diagn 2015;35:612–619.
Grande M, Jansen FA, Blumenfeld YJ, et al. Genomic microarray in fetuses with increased nuchal translucency and normal karyotype: a systematic review and meta-analysis. Ultrasound Obstet Gynecol 2015;46:650–658.
Neufeld-Kaiser WA, Cheng EY, Liu YJ. Positive predictive value of non-invasive prenatal screening for fetal chromosome disorders using cell-free DNA in maternal serum: independent clinical experience of a tertiary referral center. BMC Med 2015;13:129.
Kalousek DK, Pantzar T, Tsai M, et al. Pathology of abortion: chromosomal and genetic correlations. Monogr Pathol 1991; 33: 228–256.
Malvestiti F, Agrati C, Grimi B, et al. Interpreting mosaicism in chorionic villi: results of a monocentric series of 1001 mosaics in chorionic villi with follow-up amniocentesis. Prenat Diagn 2015;35:1117–1127.
Wallerstein R, Misra S, Dugar RB, Alem M, Mazzoni R, Garabedian MJ. Current knowledge of prenatal diagnosis of mosaic autosomal trisomy in amniocytes: karyotype/phenotype correlations. Prenat Diagn 2015;35:841–847.
Eggermann T, Soellner L, Buiting K, Kotzot D. Mosaicism and uniparental disomy in prenatal diagnosis. Trends Mol Med 2015;21:77–87.
Robinson WP, Peñaherrera MS, Jiang R, et al. Assessing the role of placental trisomy in preeclampsia and intrauterine growth restriction. Prenat Diagn 2010;30:1–8.
Sudmant PH, Mallick S, Nelson BJ, et al. Global diversity, population stratification, and selection of human copy-number variation. Science 2015;349:aab3761.
Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay. Nat Genet 2011;43:838–846.
Jensen TJ, Dzakula Z, Deciu C, van den Boom D, Ehrich M. Detection of microdeletion 22q11.2 in a fetus by next-generation sequencing of maternal plasma. Clin Chem 2012;58:1148–1151.
Srinivasan A, Bianchi DW, Huang H, Sehnert AJ, Rava RP. Noninvasive detection of fetal subchromosome abnormalities via deep sequencing of maternal plasma. Am J Hum Genet 2013;92:167–176.
Zhao C, Tynan J, Ehrich M, et al. Detection of fetal subchromosomal abnormalities by sequencing circulating cell-free DNA from maternal plasma. Clin Chem 2015;61:608–616.
Helgeson J, Wardrop J, Boomer T, et al. Clinical outcome of subchromosomal events detected by whole-genome noninvasive prenatal testing. Prenat Diagn 2015;35:999–1004.
Guex N, Iseli C, Syngelaki A, et al. A robust second-generation genome-wide test for fetal aneuploidy based on shotgun sequencing cell-free DNA in maternal blood. Prenat Diagn 2013;33:707–710.
Bayindir B, Dehaspe L, Brison N, et al. Noninvasive prenatal testing using a novel analysis pipeline to screen for all autosomal fetal aneuploidies improves pregnancy management. Eur J Hum Genet 2015;23:1286–1293.
Grati FR, Grimi B, Frascoli G, et al. Confirmation of mosaicism and uniparental disomy in amniocytes, after detection of mosaic chromosome abnormalities in chorionic villi. Eur J Hum Genet 2006;14:282–288.
Grati FR, Molina Gomes D, Ferreira JC, et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn 2015;35:801–809.
Grati FR, Malvestiti F, Ferreira JC, et al. Fetoplacental mosaicism: potential implications for false-positive and false-negative noninvasive prenatal screening results. Genet Med 2014;16:620–624.
Snyder MW, Simmons LE, Kitzman JO, et al. Copy-number variation and false positive prenatal aneuploidy screening results. N Engl J Med 2015;372:1639–1645.
Brady P, Brison N, Van Den Bogaert K, et al. Clinical implementation of NIPT - technical and biological challenges. Clin Genet 2016;89:523–530.
Acknowledgements
We thank Franz Binkert, Peter Kuhn, Alexandre Reymond, and Simon Rothen for discussions, the diagnostic team members for their devoted daily routine commitment, namely, Erika Duval, Ursula Weber, Ruth Schweizer, Antonio Chiacchio, Marion Krüger, Marco Belfiore, Nathalie Roussy, Emina Sunj, Daniel Robyr, Charlotte Sillacci, Cécile Deluen, Julien Schira, Nadine Vincent, Cristel Busca, Vianney Frigard, Marta Cotado, and Loïc Baerlocher, as well as the informatics team members, including Yves Babitch, Flavien Ombelli, and Francis Perrelet.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
(ZIP 1582 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Pescia, G., Guex, N., Iseli, C. et al. Cell-free DNA testing of an extended range of chromosomal anomalies: clinical experience with 6,388 consecutive cases. Genet Med 19, 169–175 (2017). https://doi.org/10.1038/gim.2016.72
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.72
Keywords
This article is cited by
-
Clinical outcomes of screen-positive genome-wide cfDNA cases for trisomy 20: results from the global expanded NIPT Consortium
Molecular Cytogenetics (2024)
-
Psychological impact of additional findings detected by genome-wide Non-Invasive Prenatal Testing (NIPT): TRIDENT-2 study
European Journal of Human Genetics (2024)
-
Phenotypic findings and pregnancy outcomes of fetal rare autosomal aneuploidies detected using chromosomal microarray analysis
Human Genomics (2022)
-
Prenatal diagnosis of trisomy 8 mosaicism, initially identified by cffDNA screening
Molecular Cytogenetics (2022)
-
Non-invasive prenatal testing leading to a maternal diagnosis of Charcot–Marie–Tooth neuropathy
Journal of Human Genetics (2020)
