
Abstract
Purpose:
To investigate variants of uncertain significance (VUS) in BRCA1 and BRCA2, we assessed the multifactorial posterior probability of VUS in BRCA1 and BRCA2 and compared these analyses with interpretations according to the recently released American College of Medical Genetics and Genomics (ACMG) standards and guidelines.
Methods:
The analysis involved 715 Korean patients with breast cancer. The multifactorial probability of a VUS was analyzed using the prior probability and combined likelihoods of personal and family history, the pathologic profile of the breast cancer, and co-occurrence with pathogenic variants. Results were compared with those obtained according to the ACMG standards/guidelines.
Results:
Sixteen VUS from 51 BRCA1 VUS carriers and 28 VUS from 62 BRCA2 VUS carriers were analyzed. There was a slight agreement between the two analyses, with a kappa value of 0.14 (95% confidence interval (CI) = −0.34 to 0.62) for the BRCA1 VUS and a kappa value of 0.17 (95% CI = −0.10 to 0.49) for the BRCA2 VUS.
Conclusion:
We propose that genetic counseling should be based on the concordant results between these two analyses. When discrepancies are found, those variants are still considered VUS and careful counseling should be provided.
Genet Med 18 12, 1250–1257.
Similar content being viewed by others

Introduction
Accurate interpretation of BRCA1 and BRCA2 variants is critical for risk assessment, prevention strategies, and, by extension, treatment with, for example, poly (ADP-ribose) polymerase inhibitors, which induce synthetic lethality in BRCA-deficient cells1,2,3 in patients with suspicion of hereditary breast and ovarian cancer syndromes. Recently, the US Food and Drug Administration concurrently approved olaparib monotherapy for the treatment of patients with deleterious or suspected deleterious germline BRCA1 or BRCA2 mutated advanced ovarian cancer and the companion diagnostic test for determining the pathogenicity of their BRCA1 and BRCA2 genes. When the pathogenicity of variants is being determined, the presence of variants of uncertain significance (VUS), which have insufficient evidence to be classified as either pathogenic or nonpathogenic at the time of the test, has caused problems in clinical decision making. Therefore, minimizing the proportion of BRCA1 or BRCA2 VUS (currently approximately 2–15%4) when using standardized interpretation of variants has become a crucial issue.
Recently, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology released updated standards and guidelines for the interpretation of sequence variants, in which five classifications of variants using typical types of evidence (e.g., population data, computational data, functional data, segregation data) were recommended: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign.5 These categories are intended to be applied to all genes and in different laboratory settings. The five categories are based on the relative weightings of typical types of variant evidence.
Before the release of the ACMG standards/guidelines, the International Agency for Research on Cancer (IARC) Unclassified Genetic Variants Working Group proposed a system consisting of five classes of variants to describe the results of genetic testing for cancer susceptibility. And this working group encouraged the use of the probability of being pathogenic6,7: class 5, pathogenic, >0.99 posterior probability; class 4, likely pathogenic, 0.95–0.99; class 3, uncertain, 0.05–0.949; class 2, likely neutral, 0.001–0.049; and class 1, neutral, <0.001. This multifactorial probability-based model for assessing the clinical relevance of VUS in BRCA1 and BRCA2 genes has statistically quantitative characteristics based on the clinical relevance of breast or ovarian cancers.8,9,10,11,12,13 Lindor et al.8 reviewed this model and analyzed specific BRCA1 and BRCA2 VUS for reclassification.
For the interpretation of genetic variants, the evidence and analytic methods applied differ between multifactorial posterior probability analyses and the ACMG standards/guidelines. To investigate VUS in BRCA1 and BRCA2, we compared these two methods of analysis in the present study.
Materials and Methods
Subject and variant analysis
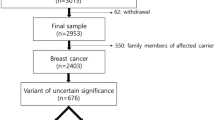
The institutional review board of Samsung Medical Center approved the study protocol (IRB No. SMC 2015-04-083). The study comprised all consecutive patients (aged ≥18 years) who underwent whole BRCA1/2 gene testing (using direct sequencing with or without multiple ligation-dependent probe amplification) between September 2001 and May 2014 at the Department of Laboratory Medicine of Samsung Medical Center in Seoul, South Korea ( Figure 1 ). Of these, Korean patients with breast cancer at Samsung Medical Center were enrolled because their clinical records were accessible. Exclusion criteria were as follows: cases requested from an external hospital, patients of non-Korean ethnicity, and patients or individuals with other diseases or conditions. We reviewed all variants of BRCA1 and BRCA2 genes in the enrolled patients. The personal and family cancer history of Korean patients with breast cancer were compared with those of European populations (Supplementary Table S1 online), and their molecular phenotypes were compared with those of other populations (Supplementary Table S2 online). A multifactorial probability-based model was developed for analysis of VUS, and ACMG released standards/guidelines for all sequence variants (not limited to VUS). The former analysis did not provide criteria such as population data (cutoff of minor allele frequency) to differentiate between benign and VUS. To compare the two analyses, we initially applied stand-alone evidence (≥5% of minor allele frequency in the 1000 Genomes Project, the Exome Sequencing Project, or the Exome Aggregation Consortium database (ExAC)) of the benign impact of the ACMG standards/guidelines on classification of benign variants. Therefore, all variants were initially classified as pathogenic (nonsense, frameshift, sequence alternation at a splice junction, initiation codon, or deletion or duplication of exons) or benign variants (≥5% of minor allele frequency in the 1000 Genomes Project, the Exome Sequencing Project, or ExAC), and the remainder were classified as VUS ( Figure 1 ). VUS were considered absent or rare (<5% of allele frequency) in these three databases and included missense variants, silent variants, or intronic variants far from the intron–exon boundary. Overall, we classified patients into the following seven groups: carriers of BRCA1 pathogenic variants (BRCA1+), carriers of BRCA2 pathogenic variants (BRCA2+), carriers of both BRCA1 and BRCA2 pathogenic variants (BRCA1/2+), neutral (with only benign variants or wild-type), BRCA1 VUS carriers, BRCA2 VUS carriers, and carriers of VUS in both BRCA1 and BRCA2. Prior to multifactorial posterior probability analyses, we analyzed the likelihood of personal and family cancer history and pathologic profiles to assess VUS pathogenicity. Then, individuals with variants of likely benign probability or possible ambiguous interpretation due to multiple VUS were eliminated in a multifactorial probability analysis. Exclusion criteria were as follows: individuals with silent variants with no prediction of splicing impact using in silico predictive algorithms, individuals with intronic variants far from the intron–exon boundary with no prediction of alternative splicing impact, individuals who had two or more VUS in each BRCA1 or BRCA2, individuals with VUS in both BRCA1 and BRCA2, or individuals with pathogenic variants in both BRCA1 and BRCA2.

Analysis workflow. Likelihoods of personal/family history and pathologic profiles were analyzed in the BRCA1+, BRCA2+, and neutral groups, and are indicated by a striped rectangle with rounded corners. BRCA1 variants of uncertain significance (VUS) and BRCA2 VUS are indicated by a gray rectangle with rounded corners using our strategies. Cases with both pathogenic variants and VUS in each BRCA1 or BRCA2 were considered BRCA1+ or BRCA2+ in this analysis workflow. BRCA1+, carriers of BRCA1 pathogenic variants; BRCA2+, carriers of BRCA2 pathogenic variants; BRCA1/2+, carriers of both BRCA1 and BRCA2 pathogenic variants; Neutral, with only benign variants or the wild-type; BRCA1 VUS, BRCA1 VUS carriers; BRCA2 VUS, BRCA2 VUS carriers; BRCA1/2 VUS, carriers of VUS in both BRCA1 and BRCA2.
Personal and family history
After a review of all medical charts of enrolled patients, we analyzed the likelihood ratios (LRs) of personal history and family history between the BRCA1+ or BRCA2+ groups and the neutral group. In a previous study,9 the LRs of personal and family history were calculated by fitting a logistic regression model. However, when one individual fit multiple categories, multiplication from multiple personal categories or family history categories could result in overestimation of the magnitude of the LRs due to the nonindependence of factors associated with personal and family history. In addition, our data did not include any cases in reference categories for multifactorial logistic regression because the present study comprised only Korean patients with breast cancer who underwent BRCA1/2 analysis—there were no subjects who were BRCA1+ or BRCA2+ but without breast cancer, nor were there subjects with neutral variants but without breast cancer. Therefore, we estimated a simple interval LR, which considers both personal and family history. The following 10 categories were used in the analysis: unilateral breast cancer; age at diagnosis of breast cancer (<40 years) and number of family members with breast, ovarian, prostate, or pancreatic cancer (0, 1, 2, and ≥3); unilateral breast cancer; age at diagnosis of breast cancer (≥40 years); and number of family members with related cancers (0, 1, 2, and ≥3); bilateral breast cancers or a combination of breast and ovarian cancers (breast/ovarian cancer), regardless of age at onset; and number of family members with related cancer (0 and ≥1). A first- or second-degree family history of cancer was defined considering identity by descent and was assessed as twice the number of first-degree relatives with cancer plus the number of second-degree relatives with cancer. If patients had a family history of cancer, as specified above, on both their paternal and maternal sides, then the side with the greater number of family members with cancer was used in the analysis.
Pathologic profile
We reviewed the phenotypic findings of breast cancers in the BRCA1+/BRCA2+ and neutral groups. Pathologic diagnosis, histologic grade (Bloom-Richardson grade), and immunohistochemistry profiles—including estrogen receptor, progesterone receptor, HER2, Ki-67, cytokeratins 5/6, and epidermal growth factor receptor—were reviewed. For HER2, an immunohistochemistry score of 0 or 1+ was classified as negative, 2+ was equivocal, and 3+ was positive, according to the recommendations of the American Society of Clinical Oncology and the College of American Pathologists.14 Additional HER2 gene amplification testing of tumors equivocal (immunohistochemistry score 2+) for HER2 protein expression was performed using fluorescence in situ hybridization (PathVysion HER2 DNA Probe kit, Abbott Molecular, Des Plaines, IL) or silver-enhanced in situ hybridization (INFORM HER2 Dual ISH assay, Ventana Medical Systems, Tucson, AZ). For Ki-67, quantitative image analysis (i-Solution Delta, InnerView, Gyeonggi-do, South Korea) was performed. Next, we classified the breast cancers of BRCA1+ and BRCA2+, and neutral groups were classified into the following four molecular subtypes: luminal A, luminal B (HER2-, HER2+), HER2 overexpression, and Basal-like.15 A cutoff value of 14% for Ki-67 was used to differentiate the luminal A from luminal B (HER2-) subtypes.16 The interval LRs of the four molecular subtypes combined with histologic grade was analyzed between the BRCA1+ or BRCA2+ and neutral groups.
Co-occurrence with pathogenic variants
Analysis of co-occurrence was conducted using the LR formulation described by Easton et al.9 In this formulation, we applied the overall frequency of BRCA1 or BRCA2 pathogenic variants derived in the current study. The probability of being a compound heterozygote for two pathogenic mutations in the BRCA1 gene was applied similarly for BRCA2 because a biallelic deleterious BRCA1 mutation in a woman with early-onset ovarian cancer was reported recently.17
Multifactorial probability analysis
To determine the prior probability of causality for a particular VUS, we used the Align-GVGD (Grantham variation/Grantham deviation) algorithm (http://agvgd.iarc.fr/agvgd_input.php) and applied prior probability according to the Align-GVGD grade as reported previously.8,18 To determine the combined likelihood of pathogenicity for a particular VUS, LRs for personal and family history, pathologic profile of breast cancer, and co-occurrence with pathogenic variants were multiplied together. The multifactorial probability of a VUS being pathogenic was analyzed using prior probability and combined likelihood based on Bayes theorem.8 Because an LR of 0.00 in the three individual likelihood analyses cannot be used in a Bayesian calculation, we assigned an LR of 0.10 instead of 0.00. In addition, when there were multiple individuals with the same VUS in those likelihood analyses, the individual LRs were multiplied together. The analyzed probability was classified based on the IARC criteria outlined in a previous study.7
Interpretation of VUS according to the ACMG standards/guidelines
For VUS analysis using the ACMG standards/guidelines,5 we primarily used ARUP (http://arup.utah.edu/database/BRCA/), Breast Cancer Information Core (BIC; http://research.nhgri.nih.gov/bic/), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php), the Leiden Open Variation Database (LOVD; http://databases.lovd.nl/shared/variants/BRCA1/unique, http://databases.lovd.nl/shared/variants/BRCA2/unique), and the UMD databases (http://www.umd.be/) for evidence of BP6 (supporting evidence of benign impact; reputable source recently reported variant as benign) or PP5 (supporting evidence of pathogenicity; reputable source recently reported variants as pathogenic) and searched for related reports (all databases accessed on 31 December 2015). For simplicity, the results of the analysis of all databases were described as follows: “5–Definitely pathogenic” in ARUP, “Class 5” in BIC, “Pathogenic” in ClinVar, “DM” in HGMD, “+ by the curator” in LOVD, and “5–Causal” in UMD as pathogenic; “4–Likely pathogenic” in ARUP, “Class 4” in BIC, “Likely pathogenic” in ClinVar, “DM?” in HGMD, “+? by the curator” in LOVD, and “4–Likely Causal” in UMD as likely pathogenic; “3–Uncertain” in ARUP, “Class 3” in BIC, “Uncertain significance” in ClinVar, “? by the curator” in LOVD, and “3–UV” in UMD as uncertain significance; “2–Likely not pathogenic or of little clinical significance” in ARUP, “Class 2” in BIC, “Likely benign” in ClinVar, “DP or DFP” in HGMD, “-? by the curator” in LOVD, and “2–Likely neutral” in UMD as likely benign; “1– Not pathogenic or of no clinical significance” in ARUP, “Class 1” in BIC, “Benign” in ClinVar, “- by the curator” in LOVD, and “1–Neutral” in UMD as benign; and “pending” or “not classified” in each database, or conflicting analysis results by multiple submitters or masters in ClinVar or LOVD, or not determined. Among these six databases, ≥50% agreement (of at least three databases) was considered evidence of BP6 or PP5. Then, we reviewed the previously published literature for analysis of variants for BS1 (strong evidence of benign impact; allele frequency is greater than expected for disorder), BS3 (strong evidence of benign impact; well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing), PS3 (strong evidence of pathogenicity; well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product), or PS4 (strong evidence of pathogenicity; the prevalence of the variants in affected individuals is significantly increased compared with the prevalence in controls). To perform an unbiased comparison of multifactorial probability-based analyses and analyses using the ACMG standards/guidelines, we excluded previous studies on multifactorial probability-based analyses for VUS as relevant references. In addition, we analyzed the difference between VUS allele frequencies of Korean breast cancer patients and Korean controls from the data reported previously. An allele frequency in controls that is significantly greater (P < 0.05) than that of breast cancer patients was considered as BS1. We used the following five in silico software packages for evidence of BP4 (supporting evidence of benign impact; multiple lines of computational evidence suggest no impact on gene or gene product) or PP3 (supporting evidence of pathogenicity; multiple lines of computational evidence support a deleterious effect on the gene or gene product): Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), PROVEAN (http://provean.jcvi.org/index.php), nsSNPAnalyzer (http://snpanalyzer.uthsc.edu/), and MutationTaster (http://www.mutationtaster.org/). In addition, previously reported predictive studies without in vitro or in vivo functional experiments were considered evidence of BP4 or PP3. For simplicity, results “probably damaging” in PolyPhen-2, “affect protein function” in SIFT, “deleterious” in PROVEAN, “disease” in nsSNPAnalyzer, and “disease-causing” in MutationTaster were indicated as deleterious. In addition, the results “benign” in PolyPhen-2, “tolerate” in SIFT, “neutral” in PROVEAN, “neutral” in nsSNPAnalyzer, and “polymorphism” in MutationTaster were indicated as neutral. For in silico analyses and predictive studies, ≥80% agreement for the prediction was considered evidence of BP4 or PP3.
Statistical analysis
Statistical analysis was performed using MedCalc Statistical Software, version 15.2.2 (MedCalc Software, Ostend, Belgium; http://www.medcalc.org). For the likelihood analyses, interval LRs with 95% CI were calculated. The differences between VUS allele frequencies of patients and controls were analyzed using the Fisher exact test. P<0.05 was regarded as indicative of statistical significance. We calculated κ interrater agreement values with 95% CI between classifications of VUS by a multifactorial probability-based model and those according to the ACMG standards/guidelines. Because the two analyses used different criteria for classification, in each analysis we categorized class 1/2, class 3, and class 4/5 according to the IARC classification, and we categorized benign/likely benign, uncertain significance, and likely pathogenic/pathogenic according to the ACMG standards/guidelines. The κ values were interpreted as follows: 0.0–0.20, slight; 0.21–0.40, fair; 0.41–0.60, moderate; 0.61–0.80, substantial; and 0.81–1.0, almost perfect agreement.
Results
From a total of 1,094 patients, the clinical data of 715 Korean patients with breast cancer were obtained. After application of the exclusion criteria, 61 BRCA1+, 70 BRCA2+, 404 neutral, 46 BRCA1 VUS carriers (51 BRCA1 VUS carriers including 5 with both pathogenic variants and VUS in BRCA1), and 55 BRCA2 VUS carriers (62 BRCA2 VUS carriers including 7 cases with both pathogenic variants and VUS in BRCA1) were included in the final data set ( Figure 1 ).
Comparing the BRCA1+ and neutral groups according to personal/family history (Supplementary Table S3 online), BRCA1+ was associated with a significantly increased risk of unilateral breast cancers with onset at age younger than 40 years, and was increased in patients with two family members with related cancers (LR = 2.03; 95% CI = 1.01–4.09) or three or more family members with related cancers (LR = 12.66; 95% CI = 3.26–49.21). In addition, bilateral breast cancers or breast/ovarian cancers with a family history of related cancers were also associated with a 2.53-fold increased LR (95% CI = 1.02–6.26). The BRCA2+ group exhibited a significantly increased likelihood in unilateral breast cancers with an onset age of younger than 40 years and with two family members who have related cancers (LR = 2.19; 95% CI = 1.15–4.17) compared to the neutral group. The likelihood was also significantly increased in bilateral breast cancers or breast/ovarian cancers with any family history (LR = 5.19; 95% CI = 2.63–10.24).
The pathologic profiles of the BRCA1+ or BRCA2+ and neutral groups were analyzed according to the four molecular subtypes combined with histologic grade. The BRCA1+ group had a significantly increased likelihood (LR = 5.01; 95% CI = 3.75–6.70) of breast cancer of the basal-like subtype and grade 3, whereas the BRCA2+ group had a significantly increased likelihood (LR = 1.80; 95% CI = 1.15–2.85) of breast cancer of the luminal B subtype and grade 2.
The multifactorial probability of VUS in BRCA1 and BRCA2 was analyzed ( Tables 1 and 2 , Supplementary Tables S4 and S5 online). Sixteen VUS from a total of 51 BRCA1 VUS carriers were analyzed as follows: IARC class 1 for 6 variants; class 2 for 6 variants; and class 3 for 4 variants. Analysis of the 28 VUS from 62 BRCA2 VUS carriers indicated that 5 variants were class 1, 19 variants were class 2, and 4 variants were class 3. Comparing the IARC class of VUS in the present study to those reported previously8 indicated that 3 of 5 BRCA1 VUS and 2 of 7 BRCA2 VUS were of the same class. One BRCA1 VUS and four BRCA2 VUS showed differences of one class (between class 1 and class 2). Differences of two classes (between class 1 and class 3) were shown in one BRCA1 VUS and one BRCA2 VUS. These BRCA1 and BRCA2 VUS were analyzed according to the ACMG standards/guidelines ( Tables 1 and 2 , Supplementary Tables S6 and S7 online). There was a slight agreement between the two analyses, with a κ value of 0.14 (95% CI = −0.34 to 0.62) in the BRCA1 VUS a κ value of 0.17 (95% CI = −0.10 to 0.49) in the BRCA2 VUS.
Discussion
To our knowledge, this is the first study to compare two analyses of VUS in the BRCA1 and BRCA2 genes. These comparisons were equitably attempted with BRCA1 and BRCA2 VUS from Korean patients with breast cancer not involved in previous studies.8,9,10,11,12,13,19
Interestingly, we did not find more than a moderate correlation (>0.41 κ value) between the two analyses, although variants with a pathogenic impact greater than IARC class 4 or likely pathogenic according to the ACMG standards/guidelines were not evaluated. This may be because the two analyses used different types of evidence to classify the variants. For example, pathologic profiles in the multifactorial posterior probability model were absent from the ACMG standards/guidelines, whereas population data, which were easily accessible using public databases such as the 1000 Genomes Project, the Exome Sequencing Project, or ExAC or by using case–control comparison, were not considered in the multifactorial posterior analyses.
Also, in the multifactorial probability-based model, the LR or odds ratio between the BRCA1+ group or BRCA2+ group and the neutral group was essential for application of the evidence of personal/family history or pathologic profiles, and the populations used in these analyses should be ethnically matched. If these accumulated analyses are insufficient, then the multifactorial probability-based model for the interpretation of VUS would be less easily applicable in a clinical laboratory setting. Unfortunately, such integral analyses have been limited to Western populations until the present. In addition, the results of these integral analyses depend on the specific factors used in the prediction by researchers. For example, in the present study, we analyzed molecular subtypes of breast cancer by comparing tumors from BRCA1+ or BRCA2+ carriers with those from noncarriers. This assessment was distinct from that of previous studies.8,10,12 The prognoses and treatment responses of breast cancer differ according to molecular subtype.15 BRCA1+ tumors are predominantly basal-like, whereas the majority of BRCA2+ tumors are of the luminal subtype, as determined by gene expression array.20,21,22 Because it was not feasible to obtain gene expression information from individual tumors, we classified our analyzed data according to the surrogate definitions for intrinsic subtypes recommended by the St. Gallen International Breast Cancer Conference Expert Panel.15 In comparison, the ACMG standards/guidelines have been developed as general guidelines for interpretation of sequence variants by genetic tests used in clinical laboratories, and applied evidence is based primarily on previous literature and public databases. Therefore, interpretation of variants is feasible despite the lack of intermediate analyses, such as likelihood of personal/family history or pathologic profile, and is less influenced by the specific factors used in predictions.
Third, evidence of pathogenicity in the ACMG standards/guidelines is categorized into four weighted grades, from supporting to very strong, whereas individual likelihoods have equal values in the multifactorial posterior probability analysis. Sometimes cosegregation analysis can become the most influential factor for pathogenicity determination in a multifactorial posterior probability analysis, although we could not perform such an analysis due to a lack of pedigree. For example, BRCA1:c.122A>G (p.His41Arg), which has been reported previously,19 was classified as IARC class 5 because of a strong cosegregation score calculated from only two families. However, segregation analysis in the ACMG standards/guidelines is regarded as supporting evidence of pathogenicity. Identification of cosegregation in multiple affected family members and multiple families from diverse ethnic populations could be considered moderate or strong evidence.
In addition, the ACMG standards/guidelines consider critical flexibility in variant classification based on professional judgment,5 whereas IARC applies individual likelihoods to posterior probability without flexibility. Therefore, one weight grade can be changed to another using expert judgment according to the ACMG standards/guideline. In the current study, the ACMG standards/guidelines were applied strictly without any flexibility. Use of such expert judgment would probably result in greater agreement in interpretation of BRCA1 and BRCA2 VUS between the IARC and ACMG standards/guidelines.
When we analyzed variants according to the ACMG standards/guideline, several types of evidences were unclear. Therefore, they were not considered other related factors and were applied simply. Of the various population databases, ExAC contains a variety of large-scale sequencing projects. Among those, exomes from The Cancer Genome Atlas Project were included. Therefore, there was a possibility, albeit slight, of inappropriate classification of variants caused by the presence in ExAC of variants related to hereditary cancers. We primarily used six locus-specific databases to search for related literature. Secondarily, we applied the BP5 or PP6 evidence using these locus-specific databases despite the limitations, such as curation problems, nomenclature errors of variants, low accuracy, and duplication due to use of overlapping data sets. Based on the low agreement among these locus-specific databases,23 ≥50% agreement (of at least three databases) was regarded as evidence of BP6 or PP5. However, we could not apply this evidence to BRCA1 or BRCA2 VUS because of disagreement among the locus-specific databases.
As reported previously for multifactorial probability analyses and the ACMG standards/guidelines, these variant analyses are still imperfect.5,8 Above all, it is difficult to generate significant information regarding genotype–phenotype correlations because most VUS were identified in a very small number of individuals and families. In addition, the multifactorial probability analysis and the ACMG standards/guidelines have not been fully clinically validated. Therefore, the strict comparison of the two analyses in the present study is limited by the fact that it is unknown which analysis has superior precision. Furthermore, this precision could not be determined simply by the proportion of VUS or by which analysis result could show the more dramatic decline in BRCA1 or BRCA2 VUS rate.
Although a five-tier classification has been proposed by two analyses, patients would be counseled regarding cancer surveillance in the following three-tier classification: counseling as if no pathogenic variant was detected for class 1/2 or benign/likely benign, counseling based on other risk factors because class 3 or uncertain significance remains a true VUS, and counseling about full high-risk surveillance of cancer that is class 4/5 or that is likely pathogenic/pathogenic.
In conclusion, this study is the first to compare multifactorial probability analysis and the 2015 ACMG standards/guidelines for interpretation of BRCA1 and BRCA2 VUS. Based on our comparative analysis, we recommend that both multifactorial probability analysis and the ACMG standards/guidelines should be used for analysis of BRCA1 and BRCA2 VUS because of the high discordance between the two analyses and because the clinical efficacy of these analyses has not been fully evaluated. In addition, genetic counseling should be based on concordant results between the two analyses. However, in the presence of discrepancies, relevant variants should still be considered VUS until further analyses of other individuals or families with those relevant variants are performed, and careful counseling is needed.
Disclosure
The authors declare no conflict of interest.
References
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–250.
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 2011;12:852–861.
Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010;376:235–244.
Cheon JY, Mozersky J, Cook-Deegan R. Variants of uncertain significance in BRCA: a harbinger of ethical and policy issues to come? Genome Med 2014;6:121.
Richards S, Aziz N, Bale S, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
Greenblatt MS, Brody LC, Foulkes WD, et al.; IARC Unclassified Genetic Variants Working Group. Locus-specific databases and recommendations to strengthen their contribution to the classification of variants in cancer susceptibility genes. Hum Mutat 2008;29:1273–1281.
Plon SE, Eccles DM, Easton D, et al.; IARC Unclassified Genetic Variants Working Group. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008;29:1282–1291.
Lindor NM, Guidugli L, Wang X, et al. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat 2012;33:8–21.
Easton DF, Deffenbaugh AM, Pruss D, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet 2007;81:873–883.
Chenevix-Trench G, Healey S, Lakhani S, et al.; kConFab Investigators. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res 2006;66:2019–2027.
Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ ; Breast Cancer Information Core (BIC) Steering Committee. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet 2004;75:535–544.
Spearman AD, Sweet K, Zhou XP, McLennan J, Couch FJ, Toland AE. Clinically applicable models to characterize BRCA1 and BRCA2 variants of uncertain significance. J Clin Oncol 2008;26:5393–5400.
Spurdle AB, Lakhani SR, Healey S, et al.; kConFab Investigators. Clinical classification of BRCA1 and BRCA2 DNA sequence variants: the value of cytokeratin profiles and evolutionary analysis–a report from the kConFab Investigators. J Clin Oncol 2008;26:1657–1663.
Wolff AC, Hammond ME, Schwartz JN, et al.; American Society of Clinical Oncology; College of American Pathologists. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol 2007;25:118–145.
Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thürlimann B, Senn HJ ; Panel members. Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol 2011;22:1736–1747.
Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009;101:736–750.
Domchek SM, Tang J, Stopfer J, et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov 2013;3:399–405.
Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat 2008;29:1342–1354.
Whiley PJ, Parsons MT, Leary J, et al. Multifactorial likelihood assessment of BRCA1 and BRCA2 missense variants confirms that BRCA1:c.122A>G(p.His41Arg) is a pathogenic mutation. PLoS One 2014;9:e86836.
Waddell N, Arnold J, Cocciardi S, et al.; kConFab Investigators. Subtypes of familial breast tumours revealed by expression and copy number profiling. Breast Cancer Res Treat 2010;123:661–677.
Jönsson G, Staaf J, Vallon-Christersson J, et al. Genomic subtypes of breast cancer identified by array-comparative genomic hybridization display distinct molecular and clinical characteristics. Breast Cancer Res 2010;12:R42.
Larsen MJ, Kruse TA, Tan Q, et al. Classifications within molecular subtypes enables identification of BRCA1/BRCA2 mutation carriers by RNA tumor profiling. PLoS One 2013;8:e64268.
Vail PJ, Morris B, van Kan A, et al. Comparison of locus-specific databases for BRCA1 and BRCA2 variants reveals disparity in variant classification within and among databases. J Community Genet 2015;6:351–359.
Acknowledgements
This study was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A120030).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Tables
(DOC 631 kb)
Rights and permissions
About this article

Cite this article
Park, K., Cho, E., Nam, S. et al. Comparative analysis of BRCA1 and BRCA2 variants of uncertain significance in patients with breast cancer: a multifactorial probability-based model versus ACMG standards and guidelines for interpreting sequence variants. Genet Med 18, 1250–1257 (2016). https://doi.org/10.1038/gim.2016.39
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.39
