
Abstract
Purpose:
Approximately two-thirds of patients with the lysosomal storage disease mucopolysaccharidosis II have progressive cognitive impairment. Intravenous (i.v.) enzyme replacement therapy does not affect cognitive impairment because recombinant iduronate-2-sulfatase (idursulfase) does not penetrate the blood–brain barrier at therapeutic concentrations. We examined the safety of idursulfase formulated for intrathecal administration (idursulfase-IT) via intrathecal drug delivery device (IDDD). A secondary endpoint was change in concentration of glycosaminoglycans in cerebrospinal fluid.
Methods:
Sixteen cognitively impaired males with mucopolysaccharidosis II who were previously treated with weekly i.v. idursulfase 0.5 mg/kg for ≥6 months were enrolled. Patients were randomized to no treatment or 10-mg, 30-mg, or 1-mg idursulfase-IT monthly for 6 months (four patients per group) while continuing i.v. idursulfase weekly.
Results:
No serious adverse events related to idursulfase-IT were observed. Surgical revision/removal of the IDDD was required in 6 of 12 patients. Twelve total doses were administrated by lumbar puncture. Mean cerebrospinal fluid glycosaminoglycan concentration was reduced by approximately 90% in the 10-mg and 30-mg groups and approximately 80% in the 1-mg group after 6 months.
Conclusions:
These preliminary data support further development of investigational idursulfase-IT in MPS II patients with the severe phenotype who have progressed only to a mild-to-moderate level of cognitive impairment.
Genet Med 18 1, 73–81.
Similar content being viewed by others

Introduction
Mucopolysaccharidosis II (MPS II, Hunter syndrome; OMIM 309900) is an X-linked lysosomal storage disease caused by a deficiency in the enzyme iduronate-2-sulfatase (I2S), which is required for the catabolism of glycosaminoglycans (GAG).1 Accumulation of GAG in cells, tissues, and organs produces multi-systemic, progressive disease.2,3 Approximately two-thirds of patients have the severe phenotype, characterized by progressive cognitive decline.4
Weekly infusions of idursulfase, recombinant human I2S (Elaprase, Shire, Lexington, MA), have been shown to improve some somatic manifestations in MPS II patients,5,6 but the enzyme does not penetrate the blood–brain barrier at therapeutic doses or alter cognitive decline in severe disease.7 Animal studies have demonstrated that intrathecally administered idursulfase can penetrate into brain tissue.8 Therefore, the present phase I/II trial (NCT00920647) studied the direct administration of investigational recombinant I2S into the cerebrospinal fluid (CSF) as a means of delivering the drug to the central nervous system (CNS). The investigational product, idursulfase-IT, is a formulation of recombinant human I2S for intrathecal (IT) administration into CSF using an IT drug delivery device (IDDD). The primary study objective was to determine the safety and tolerability of ascending doses of idursulfase-IT administered via IDDD once monthly for 6 months to MPS II patients with cognitive impairment who were concurrently receiving intravenous (i.v.) idursulfase. This novel approach is supported by existing knowledge of the leptomeningeal space and molecule transfer from CSF into the brain parenchyma.8,9
Materials and Methods
Patients
Patients with MPS II were recruited between June 2009 and March 2012 and enrolled at the University of North Carolina-Chapel Hill, Chapel Hill, North Carolina, USA, and Birmingham Children’s Hospital, Birmingham, UK. Included patients were males who were between 3 and 18 years of age who tolerated ≥6 months of treatment with 0.5 mg/kg weekly i.v. idursulfase before screening. Included patients had evidence of MPS II–related cognitive impairment on formal neurodevelopmental assessment at screening. Initially, cognitive impairment was defined as an overall intelligence quotient (IQ) that was ≥2 but ≤3 SDs below the mean of the healthy population. Alternatively, a decline of at least 1 SD in IQ score was permitted as evidence of cognitive impairment, provided the patient’s IQ remained ≤3 SD from the mean of the healthy population.10,11 Because of difficulties with initial enrollment with these cognitive impairment criteria, and given that the primary endpoint of the study was safety and tolerability, these criteria were amended in the protocol several times over the course of the study. The final inclusion criteria for cognitive impairment were an IQ score ≤77 points or evidence of an IQ score decline of ≥1 SD (15 IQ points) but ≤2 SD (30 IQ points) from a previous neurodevelopmental assessment, with a duration of neurologic involvement between 3 and 36 months. During the course of the trial, all patients met the first of the final inclusion criterion for cognitive impairment (IQ score ≤77 points); no patient was actually enrolled by means of the second of the final inclusion criteria (IQ score decline).
Exclusion criteria included the following: clinically significant CNS involvement unrelated to MPS II; opening CSF pressure >30.0 cm of H2O; CNS shunt; poorly controlled seizure disorder; hematopoietic stem cell transplantation; blood product transfusions within 90 days of screening; and skeletomuscular/spinal abnormalities or other contraindications for the surgical implantation of the IDDD.
Investigational product and intrathecal drug delivery device
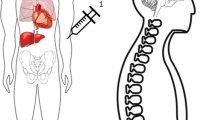
Idursulfase-IT is a sterile, isotonic solution of recombinant human I2S manufactured in single-use vials for IT administration via an indwelling IDDD (PORT-A-CATH II Low Profile Intrathecal Implantable Access System; Smiths Medical, St. Paul, MN). The dosing solution was provided at 50 mg/ml and diluted as needed in preservative-free saline. The IDDD implant was performed according to the instructions for use, with a lumbar incision to allow for the insertion of the catheter at the level of L4–L5 and a second incision on the lower ribs to allow for the implant of the access port. The catheter tip was threaded up the spinal canal to the mid-thoracic level. During access, ~7 ml of CSF was removed. The first milliliter was discarded, with the subsequent volume divided into aliquots for analysis. A flush of ≥2 ml of preservative-free saline was recommended after the dose was administered, but typically additional saline was flushed to compensate for the total volume of CSF withdrawn. The process of drug administration and flush by slow bolus took ~2–5 min.
Study design
The study was a randomized, open-label study approved by the relevant institutional review boards and conducted in compliance with the Declaration of Helsinki, relevant institutional review board regulations, and International Conference on Harmonisation Good Clinical Practice guidelines. A placebo control was not considered because of an unacceptable risk/benefit ratio associated with IDDD placement and IT administration of an inactive comparator. Written informed consent was obtained from parents or legal guardians.
Prospectively generated permuted block randomization12 using four blocks of size four each and an allocation ratio of 3:1 within each block (treatment:no treatment) was used to create three dose cohorts of five to six patients each. In each dose cohort, four patients were randomized to IDDD implantation plus administration of idursulfase-IT once monthly (every 28 ± 7 days) for 6 months, and in each dose cohort one or two patients were randomized to no treatment. Those randomized to treatment in the first, second, and third cohorts received 10-mg, 30-mg, and 1-mg idursulfase-IT monthly, respectively. In total, 16 patients were randomized—four IT-treated patients per dose group and four patients in the no treatment group.
The IDDDs were implanted in treated patients at week 1, followed by 2 weeks of recovery. The first dose of idursulfase-IT was given at week 3. CSF samples were drawn immediately before administration of each idursulfase-IT dose. For the first four doses, idursulfase-IT was administered only after CSF analysis confirmed a white blood cell (WBC) count <30 WBC/mm3, requiring ~20–40 min between CSF sampling and dose administration. If the IDDD became nonfunctional or its use was precluded, then CSF sampling and/or dosing by lumbar puncture were permitted.
The original dose-escalation study design specified the evaluation of idursulfase-IT at dose levels of 10, 30, and 100 mg. After preliminary data showed a similar pharmacodynamic response in both the 10-mg and 30-mg groups, the protocol was amended, replacing the 100-mg dose with a 1-mg dose. Therefore, we present our findings here in order of the 10-mg, 30-mg, and 1-mg groups to reflect the protocol change.
All patients continued to receive 0.5 mg/kg i.v. idursulfase weekly. At least 48 hours elapsed between idursulfase-IT dosing and subsequent i.v. idursulfase dosing.
Endpoints and assessments
The primary endpoint was safety and tolerability. Assessments included adverse events (AEs), 12-lead electrocardiogram (ECG) findings, complete blood count, serum chemistries, CSF glucose, protein, red blood cell count and WBC count including a differential, and anti-I2S antibodies in CSF and serum. Treatment-emergent adverse events (TEAEs) were defined as all AEs occurring between the time of randomization and the last follow-up contact at 30 ± 7 days after the end of this 6-month study. All eligible patients continued on to the extension study, the results of which will be reported separately. TEAEs were graded for severity from mild (grade 1) to death (grade 5).13 Serious adverse events (SAEs) were any TEAEs that were life-threatening or resulted in death, inpatient hospitalization, prolongation of existing hospitalization, or persistent or significant disability/incapacity.
All antibody testing was performed by a Shire-designated contract research organization (Pharmaceutical Product Development, LLC, Richmond, VA). Anti-I2S antibodies were detected by a bridging electrochemiluminescent immunoassay using the Meso Scale Discovery technology platform (Meso Scale Diagnostics, LLC, Rockville, MD). Biotin-idursulfase was used as the capture reagent, and sulfo-tagged idursulfase was used as the detection reagent. Positive samples were confirmed by ligand-competition assay and then further tested for the antibody titer and neutralizing activity. Neutralizing activity was detected by an I2S enzymatic activity assay using 4-methylumbelliferyl-sulfate as substrate. Titer was defined as the reciprocal of the highest sample dilution (including minimum required dilution) with a positive result.
The study’s secondary endpoints, summarized by dose group, included pharmacokinetic parameters of I2S in serum and CSF after the administration of idursulfase-IT and the change in CSF and urinary GAG concentrations from baseline. The quantification of I2S in serum and CSF was performed by Shire using an enzyme-linked immunosorbent assay as previously described.14 The quantification of total CSF GAG concentration was determined by Shire using a thrombin activity assay with chromogenic substrate S-2238 (Chromogenix, Bedford, MA). CSF samples were pre-incubated with a fixed amount of human heparin cofactor II (Haematologic Technologies, Essex Junction, VT), then incubated with a fixed amount of human α-thrombin (Enzyme Research Laboratories, South Bend, IN) and 0.5 mmol/l S-2238 in assay buffer. GAG in CSF samples binds to heparin cofactor II, which in turn accelerates thrombin inactivation. The GAG concentration in CSF samples was calculated from a calibration curve prepared from dermatan sulfate (GAG-DS01, Iduron, Manchester, UK) included in the same assay. The quantification of urinary GAG was performed by Cambridge Biomedical (Boston, MA) using a Blyscan Glycosaminoglycan Assay kit (Biocolor Life Science Assays, Carrickfergus, UK), and results were normalized to urine creatinine.
Blood samples for pharmacokinetic assessments were drawn at week 3 (first idursulfase-IT dose) and week 23 (last idursulfase-IT dose). For the analysis of the serum enzyme pharmacokinetics after intrathecal administration of idursulfase-IT, blood samples were initially drawn within 15 min before idursulfase-IT administration (pre-injection baseline), and then at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 hours after administration. Later, time points of 30 and 36 hours after administration were added by protocol amendment because early data indicated that the appearance of enzyme in serum was slow and maintained. To analyze serum pharmacokinetics after i.v. administration of idursulfase, blood samples were drawn at pre-infusion baseline, then at 0.5, 1, 1.5, 2, 2.5, and 3 hours during the infusion, and at 3.5, 4, 5, 6, 7, 9, 11, and 24 hours after the infusion. CSF samples from treated patients were collected via IDDD or lumbar puncture immediately before each administration of idursulfase-IT and at week 27 (30 ± 7 days after the sixth dose of idursulfase-IT). For patients assigned to no treatment, CSF samples were collected via lumbar puncture at screening and week 27. Urine samples were collected up to 24 hours before each administration of idursulfase-IT and at week 27 (30 ± 7 days after the sixth dose of idursulfase-IT) for treated patients, and at screening and week 27 for no treatment patients.
Data from the intent-to-treat group, comprising all randomized patients, were used in all data analyses. Data analyses were descriptive and were performed using SAS (SAS Institute, Cary, NC) version 9.3.
Results
Study population
Demographics and baseline characteristics are provided in Table 1 .
Treatment exposure
Seven of 12 treated patients (58.3%) received all scheduled doses of idursulfase-IT, either by IDDD or by lumbar puncture. Four patients (33.3%), two each in the 1-mg and 30-mg groups, received five of six scheduled doses, and one patient (8.3%) in the 30-mg group received four of six scheduled doses. Device malfunction led to partial revision, total surgical revision, or removal in 6 of 12 (50%) treated patients ( Figure 1 ).

Age, genotype, and overview of dosing and IDDD-related procedures. Implant refers to the implantation of an IDDD. Total revision refers to the removal and replacement of an IDDD in its entirety (i.e., both the access port and the catheter are removed and replaced). A partial revision refers to the removal and replacement of only the access port, with the original catheter remaining in situ. Removal refers to the removal of an IDDD in its entirety without replacement during the same procedure. IDDD, intrathecal drug delivery device; LP, lumbar puncture.
Safety assessments
Treatment with idursulfase-IT appeared generally well-tolerated ( Table 2 ). There were no deaths or discontinuations. Most TEAEs were mild to moderate (grades 1–2) in severity. There were six severe (grade 3) events experienced by three patients, one in each dose group. These included device breakage, device dislocation, complication of device insertion/orientation, procedure complication, and implant site infection. There was no evidence of increasing TEAE severity or frequency with higher doses of idursulfase-IT. There were 34 TEAEs that were assessed as possibly/probably related to idursulfase-IT. Most of these fell into the “investigations” category, such as blood pressure and heart rate abnormalities, or an increased CSF protein level. Others included behavior change, vomiting, nausea, pyrexia, erythema, anxiety, and pyramidal tract syndrome.
Fourteen SAEs were experienced by 7 of 12 (58.3%) treated patients ( Table 2 ). All SAEs were so designated because hospitalization was required. Notably, 12 of 14 (85.7%) SAEs were IDDD-related (complication of device insertion, device dislocation/connection issue, device breakage/malfunction/failure, implant site infection, procedural pain, and wound dehiscence). The other two (vomiting and dehydration) were experienced by a patient who was implanted with an IDDD but had not yet received study drug. No SAEs were reported in the no treatment group.
There were no clinically meaningful changes from baseline in serum chemistry, hematology, urinalysis laboratory values, CSF laboratory values, or ECG findings over time (data not shown). There were no important differences between treated and untreated patients or between the 1-mg, 10-mg, or 30-mg dose groups. CSF WBC count was monitored throughout the study, and no evidence was found to suggest any association between CNS inflammation and idursulfase-IT. Occasional instances of transient, asymptomatic CSF leukocytosis (maximum value, 33 WBC/mm3) were noted and typically preceded IDDD failures, sometimes by several months. All episodes resolved either spontaneously or after device revision/removal.
Immunogenicity in serum
Seven of 15 evaluated patients (47%) tested anti-I2S antibody positive in serum at week 3 (measurements were taken before IT dosing for treated patients). Note that all enrolled patients in this study had received i.v. idursulfase for ≥6 months before study entry and continued receiving it throughout the study. Of the seven patients with serum antibody positivity at week 3, six patients had stable antibody titers (within ±1 step of a 1:4 serial dilution) throughout the study. The seventh patient, in the 10-mg group, showed a generally decreased serum antibody titer over time and tested antibody–negative at week 27. An eighth patient, from the no treatment group, was serum antibody–negative at baseline but tested positive at week 27.
Of the eight patients who were serum antibody–positive at any point, two patients in the 1-mg group and one patient in the no treatment group had substantial neutralizing antibody activity (neutralizing antibody titer ≥800) that remained stable throughout the study. Weak neutralizing activity (neutralizing antibody titer <800) was seen at least once during the study for three of eight antibody-positive patients, one from each idursulfase-IT dose group. The remaining two patients, both in the no treatment group, had no detectable neutralizing antibody activity at any time during the study.
Immunogenicity in cerebrospinal fluid
Before the first exposure to idursulfase-IT, anti-I2S antibodies were detected in CSF for 6 of 15 patients (40%). The observed CSF antibody titers appeared largely stable (within ±2 steps of a 1:2 serial dilution) throughout the study. The corresponding serum samples for these six patients also tested antibody-positive; serum antibody titers were typically much higher relative to the corresponding CSF antibody titers. The highest titers were observed in patient 12 (1-mg group), who had a large deletion in the I2S gene and who had a serum antibody titer of 655,360 and CSF antibody titer of 5,120 before idursulfase-IT exposure. He had the least pronounced CSF GAG response ( Figure 3 ).

Observed GAG concentrations in CSF for individual patients by scheduled study week visit and treatment group: (a) 10 mg, (b) 30 mg, (c) 1 mg, and (d) No-Treatment. CSF, cerebrospinal fluid; GAG, glycosaminoglycan.
Of the patients who had no CSF antibodies to idursulfase at study start, none subsequently developed them. One patient in the no treatment group did not have sufficient CSF volume at baseline for immunogenicity testing but was antibody-negative in the CSF at week 27.
Pharmacokinetic parameters
After IT dosing, the 10-mg and 30-mg doses of idursulfase-IT exhibited similar serum I2S concentration-time profiles at week 23 ( Figure 2 ). Most patients had baseline I2S protein levels below the limit of quantitation, and only patients with detectable I2S protein levels are presented in Figure 2 . Limited information exists on the serum pharmacokinetic profile of idursulfase-IT at the 1-mg dose. Only two of four patients in the 1-mg idursulfase-IT group had serum I2S levels above the lower limit of quantitation, and only one of these had sufficient serum concentration-time values to derive pharmacokinetic parameters.

Mean serum iduronate-2-sulfatase concentration-time profiles at week 23. Serum samples were taken from treated patients in each of the idursulfase-IT dosing groups following administration of idursulfase-IT (n values shown are per dose level), as well as following administration of intravenous (i.v.) idursulfase ~48 h later (n value is shown together, n = 9). These data are graphed together for comparative purposes. The level of iduronate-2-sulfatase protein in serum prior to idursulfase-IT administration (t = 0) was below the lower limit of quantification in 4/9 patients; the mean value shown at t = 0 reflects only those samples with quantifiable amounts (n = 2 for the 10-mg group; n = 1 for the 30-mg group; and n = 2 for the 1-mg group, respectively). Similarly, the level of iduronate-2-sulfatase in serum prior to i.v. idursulfase administration (t = 0) was below the lower limit of quantification in 7/9 patients; the mean value shown at t = 0 reflects only those samples with quantifiable amounts (n = 1 for the 10-mg group; n = 1 for the 30-mg group; and n = 0 for the 1-mg group, respectively). CSF, cerebrospinal fluid; IT, intrathecal; i.v., intravenous.
Dose proportionality of serum I2S exposure was observed between the idursulfase-IT 1- and 10-mg groups for maximum observed serum concentration (Cmax) (7.9-fold increase), but not area under the serum concentration-time curve (AUC0–last) (4.8-fold increase). Dose proportionality of serum I2S exposure was not observed between the idursulfase-IT 10-mg and 30-mg groups for Cmax or AUC0–last. The mechanism by which intrathecally administered idursulfase-IT enters the systemic circulation is not clearly defined. One potential mechanism is that the protein is removed from the CSF through the arachnoid villi.15 Materials transverse the villi by micropinocytosis, which is a unidirectional process mediating transport from the CSF to the venous system or epidural space.
Systemic bioavailability for the 10-mg group was 44.4 ± 14.6% at week 3 and 46.8 ± 8.5% at week 23, resulting in mean equivalent i.v. doses of 0.21 ± 0.08 mg/kg at week 3 and 0.22 ± 0.05 mg/kg at week 23. Systemic bioavailability for the 30-mg group was 22.3 ± 2.2% at week 3 and 22.2 ± 14.2% at week 23, resulting in mean equivalent i.v. doses of 0.23 ± 0.06 mg/kg at week 3 and 0.21 ± 0.14 mg/kg at week 23. These observations suggest that bioavailability was consistent over time when the same dose of idursulfase-IT was administered, and that none of the patients received a systemic exposure from IT delivery of idursulfase-IT that would be equivalent to the standard prescribed dose of 0.5 mg/kg of i.v. idursulfase.
We determined the concentration of I2S in CSF prior to each idursulfase-IT dose (~30 days after the previous idursulfase-IT dose). Monthly idursulfase-IT administration at doses of 1–30 mg did not produce appreciable accumulation in the CSF (data not shown). Levels in the majority of samples were below the limit of quantification (3.13 ng/ml).
Glycosaminoglycan concentrations in cerebrospinal fluid and urine
Concentrations of GAG in CSF were evaluated as a marker for the biological activity of idursulfase-IT. CSF samples were obtained monthly, before the administration of idursulfase-IT. The observed GAG concentration at a given time point is that which is present 1 month after treatment. Because I2S is inactive in physiological fluids, such as CSF,16 observed declines in CSF GAG after idursulfase-IT administration are likely attributable to reductions in GAG concentration in tissues rather than to direct enzymatic degradation of GAG in CSF.
At week 27, 30 ± 7 days after the final idursulfase-IT dose in this study, the mean concentration (±SE) of CSF GAG declined by 90 ± 3%, 89 ± 1%, and 79 ± 5% in the 10-mg, 30-mg, and 1-mg groups, respectively. The major part of this decline was already apparent after the first dose of idursulfase-IT: 73 ± 7%; 89 ± 1%; and 59 ± 10%, respectively. In the no treatment group, the mean concentration of CSF GAG (±SE) increased by 7 ± 6% by end of study. Individually, all treated patients experienced declines in CSF GAGs from baseline, whereas all no treatment patients had stable CSF GAG levels throughout ( Figure 3 ). There was no apparent effect of idursulfase-IT on urinary GAG levels (data not shown).
Discussion
Our primary objective was to investigate the safety of idursulfase-IT administered intrathecally once monthly to children with MPS II and cognitive impairment who were receiving concurrent i.v. idursulfase once weekly. Long-term neurocognitive data from this study and an extension study are forthcoming. Here, idursulfase-IT appeared generally well-tolerated. There was no apparent relationship between the incidence or type of AEs and the dose. Although 7 of 12 treated patients experienced at least one SAE during the study, the majority (12/14) were related to IDDD malfunction. The issues associated with the device fell into two main categories: device breakage and catheter migration from the spinal canal. The high activity level in this pediatric population may have contributed to the high rate of these AEs, because young patients with MPS II and cognitive impairment often have marked hyperactivity, impulsivity, and restlessness.17,18,19 A different IDDD has been identified and implemented in subsequent studies of idursulfase-IT, to be described in future reports. Of note, there were no SAEs considered related to the study drug. There were no cases of infective meningitis. Administration of idursulfase-IT was also typically not associated with an elevated WBC count or other signs of CNS inflammation.
Idursulfase-IT did not appear to have high immunogenic potential after IT administration, although the brief 6-month duration of the study and the small number of patients preclude us from making firm conclusions in this regard. Six of 15 patients (two in the no treatment group) had anti-I2S antibodies in CSF before being exposed to idursulfase-IT. Their titers remained largely stable throughout the study, and no additional patients developed CSF antibodies during the study. These six patients also tested positive for serum antibodies, before any exposure to idursulfase-IT, at higher titers than those in CSF. This is entirely consistent with the previously published rates of antibody positivity in patients treated with idursulfase for ≥6 months.5,14 The highest titers were observed in a patient with a large deletion in the I2S gene. Patients with a complete deletion produce no endogenous enzyme, probably causing idursulfase to be viewed as a non-self antigen. Because all patients had received ≥6 months of i.v. idursulfase therapy before enrollment, the positive CSF antibody test results seen prior to idursulfase-IT exposure were likely due to movement of serum antibodies across the blood–brain barrier.20 Serum antibody titers also remained stable during the study. The only patient who developed serum antibodies during the study was in the no treatment group.
As a secondary endpoint, we assayed the concentration of CSF GAG over time. At week 27, the mean CSF GAG concentration declined by ~90% in the 10-mg and 30-mg groups and ~80% in the 1-mg group. The decline was largely apparent after the first dose of idursulfase-IT. Because CSF samples were taken 1 month after the preceding drug dose, these results indicate rapid and robust decreases in CSF GAG levels in response to idursulfase-IT that were sustained throughout the 6-month study. In contrast, the mean CSF GAG concentration for the no treatment group remained stable throughout. Although we have no direct evidence that idursulfase-IT, administered via IT bolus, penetrates the brain parenchyma in these patients, there are several indirect indications that this may be a reasonable hypothesis. First, because recombinant I2S is inactive in the CSF, these data suggest that its pharmacodynamic activity takes place in lysosomes, which requires I2S uptake from the CSF into cells. Second, animal study data have shown that recombinant I2S penetrates into the brain parenchyma after either intraparenchymal administration to a mouse model of MPS II or IT administration to healthy monkeys.8 Third, idursulfase-IT is given as a bolus injection, followed by a flush of preservative-free saline, a procedure that may help drive the drug cephalad toward the brain.9 Fourth, MPS II patients often have dilated Virchov-Robin spaces, which may result in enhanced drug delivery to the brain parenchyma.9
In the pharmacokinetic analysis, we found a mean systemic bioavailability of 22–47% for idursulfase-IT at different doses, although limited information exists at the 1-mg dose. This was not unexpected, given the known clearance mechanisms for CSF.9 However, idursulfase-IT is not expected to supplant the need for weekly i.v. idursulfase to address certain somatic features of MPS II, particularly as idursulfase-IT therapy is administered monthly, not weekly, and is not dosed based on weight. The bioavailability data support this expectation because the systemic exposure achieved in the 10-mg or 30-mg idursulfase-IT groups was much less than that obtained after a 0.5-mg/kg dose of i.v. idursulfase.
In conclusion, monthly intrathecal administration of idursulfase-IT to patients with severe MPS II appeared to be generally well-tolerated in this small study and resulted in a >70% reduction in CSF GAG concentration 1 month after the first dose of 10 or 30 mg. The IDDD did not perform as anticipated in this active (and hyperactive) pediatric population, necessitating several surgical revisions/removals during the trial and triggering the adoption of a new IDDD for subsequent studies. Our conclusions should be interpreted with caution due to the small sample size of 12 treated patients and brief 6-month duration of treatment; however, the results support the continued development of idursulfase-IT as a potential therapy for the cognitive impairment of MPS II. An extension trial is ongoing to evaluate long-term safety, immunogenicity, clinical, and cognitive outcomes associated with idursulfase-IT treatment. In addition, a phase II/III study is recruiting children with severe MPS II who have progressed only to a mild to moderate level of cognitive impairment to determine the effect of monthly administration of idursulfase-IT on neurodevelopmental status over 12 months (NCT02055118).
Disclosure
J.M. has received honoraria and educational and travel grants from Janssen R&D and Shire, and has participated in clinical trials with BioMarin Pharmaceutical and Shire. C.J.H. has received honoraria and educational and travel grants from Actelion Pharmaceuticals, BioMarin Pharmaceutical, GlaxoSmithKline, Shire, and Sanofi-Genzyme, and has participated in clinical trials with Actelion Pharmaceuticals, Amicus Therapeutics, BioMarin Pharmaceutical, GlaxoSmithKline, Shire, Sanofi-Genzyme, and Synageva BioPharma. S.V. has received honoraria and travel grants from Shire and has participated in clinical trials with Shire. S.S. has received educational and travel grants from Shire and has participated in clinical trials with Shire and BioMarin Pharmaceutical. G.A.S. has received honoraria and travel grants from and has participated in clinical trials with BioMarin Pharmaceutical and Shire. M.A.M., L.P., K.S., A.J.B., and N.W. are employees of Shire. The other authors declare no conflict of interest.
References
Bach G, Eisenberg F Jr, Cantz M, Neufeld EF. The defect in the Hunter syndrome: deficiency of sulfoiduronate sulfatase. Proc Natl Acad Sci USA 1973;70:2134–2138.
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease, vol. III. McGraw-Hill: New York, 2001:3421–3452.
Martin R, Beck M, Eng C, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008;121:e377–e386.
Young ID, Harper PS, Newcombe RG, Archer IM. A clinical and genetic study of Hunter’s syndrome. 2. Differences between the mild and severe forms. J Med Genet 1982;19:408–411.
Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med 2006;8:465–473.
Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med 2011;13:95–101.
Muenzer J, Bodamer O, Burton B, et al. The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur J Pediatr 2012;171:181–188.
Calias P, Papisov M, Pan J, et al. CNS penetration of intrathecal-lumbar idursulfase in the monkey, dog and mouse: implications for neurological outcomes of lysosomal storage disorder. PLoS One 2012;7:e30341.
Papisov MI, Belov VV, Gannon KS. Physiology of the intrathecal bolus: the leptomeningeal route for macromolecule and particle delivery to CNS. Mol Pharm 2013;10:1522–1532.
Elliott CD. Differential Ability Scales–Second Edition (DAS-II®), 2nd edn. Harcourt Assessment: San Antonio, TX, 2007.
Bayley N. Bayley Scales of Infant and Toddler Development (Bayley-III®), 3rd edn. Pearson: San Antonio, TX, 2005.
Matts JP, Lachin JM. Properties of permuted-block randomization in clinical trials. Control Clin Trials 1988;9:327–344.
The National Cancer Institute. Common Terminology Criteria for Adverse Events Version 4.0. http://evs.nci.nih.gov/ftp1/CTCAE/About.html. Accessed 15 January 2014.
Barbier AJ, Bielefeld B, Whiteman DA, Natarajan M, Pano A, Amato DA. The relationship between anti-idursulfase antibody status and safety and efficacy outcomes in attenuated mucopolysaccharidosis II patients aged 5 years and older treated with intravenous idursulfase. Mol Genet Metab 2013;110:303–310.
Laterra J, Keep R, Betz LA, Goldstein GW. Blood—brain—cerebrospinal fluid barriers. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD (eds). Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, 6th edn. Lippincott Williams & Wilkins: Philadelphia, PA, 1999:671–689.
Bielicki J, Freeman C, Clements PR, Hopwood JJ. Human liver iduronate-2-sulphatase. Purification, characterization and catalytic properties. Biochem J 1990;271:75–86.
Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis Child 1995;73:77–81.
Holt J, Poe MD, Escolar ML. Early clinical markers of central nervous system involvement in mucopolysaccharidosis type II. J Pediatr 2011;159:320–326.e2.
Muenzer J, Beck M, Eng CM, et al. Multidisciplinary management of Hunter syndrome. Pediatrics 2009;124:e1228–e1239.
Reiber H. Flow rate of cerebrospinal fluid (CSF)–a concept common to normal blood-CSF barrier function and to dysfunction in neurological diseases. J Neurol Sci 1994;122:189–203.
Acknowledgements
Clinical trial HGT-HIT-045 (NCT00920647) is funded by Shire. Editorial assistance to the authors for the preparation of the manuscript was funded by Shire and was provided by Jillian Lokere and Nicky Lowney of the Curry Rockefeller Group, LLC, Tarrytown, NY, USA. The authors received no payment for their work and confirm independence from the funding source.
The authors thank the patients and their families for their participation in the study. The authors acknowledge Margot Stein, Lynn Wegner, Pat Rausch, Heather Preiss, and Sara Richardson (University of North Carolina at Chapel Hill, Chapel Hill, NC, USA) and Shauna Kearney, Johan Horton, Shima Sinha, and Rebecca Lawrence (Birmingham Children’s Hospital NHS Foundation Trust, Birmingham, UK) for their contributions to the study. They also thank Judith Healy and Sean Seyffert of Shire, Lexington, MA, USA, for assisting in the writing of the clinical study report.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Muenzer, J., Hendriksz, C., Fan, Z. et al. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet Med 18, 73–81 (2016). https://doi.org/10.1038/gim.2015.36
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2015.36
Keywords
This article is cited by
-
Mature neurons from iPSCs unveil neurodegeneration-related pathways in mucopolysaccharidosis type II: GSK-3β inhibition for therapeutic potential
Cell Death & Disease (2024)
-
A novel mucopolysaccharidosis type II mouse model with an iduronate-2-sulfatase-P88L mutation
Scientific Reports (2023)
-
Evolving therapies in neuronopathic LSDs: opportunities and challenges
Metabolic Brain Disease (2022)
-
Die Grenzen der Enzymersatztherapien sprengen
Pädiatrie (2021)
-
Neue Therapien bei lysosomalen Speicherkrankheiten
Pädiatrie & Pädologie (2021)
