
Abstract
Purpose:
The purpose of this study was to assess the diagnostic yield of the traditional, comprehensive clinical evaluation and targeted genetic testing, within a general genetics clinic. These data are critically needed to develop clinically and economically grounded diagnostic algorithms that consider presenting phenotype, traditional genetics testing, and the emerging role of next-generation sequencing (whole-exome/genome sequencing).
Methods:
We retrospectively analyzed a cohort of 500 unselected consecutive patients who received traditional genetic diagnostic evaluations at a tertiary medical center. We calculated the diagnosis rate, number of visits to diagnosis, genetic tests, and the cost of testing.
Results:
Thirty-nine patients were determined to not have a genetic disorder; 212 of the remaining 461 (46%) received a genetic diagnosis, and 72% of these were diagnosed on the first visit. The cost per subsequent successful genetic diagnosis was estimated at $25,000.
Conclusion:
Almost half of the patients were diagnosed using the traditional approach, most at the initial visit. For those remaining undiagnosed, next-generation sequencing may be clinically and economically beneficial. Estimating a 50% success rate for next-generation sequencing in undiagnosed genetic disorders, its application after the first clinical visit could result in a higher rate of genetic diagnosis at a considerable cost savings per successful diagnosis.
Genet Med 16 2, 176–182.
Similar content being viewed by others
Introduction
The medical, psychosocial, and economic burden of genetic disorders has long been recognized; in children, these disorders account for upwards of 25% of inpatient admissions, as well as increased outpatient visits.1,2,3 Despite this high disease burden, many genetic conditions are intractable to diagnostic evaluation, despite recent advances in molecular laboratory investigations.2,4,5 The diagnostic process employed by most medical geneticists is tiered, with clinical assessment followed by sequential laboratory testing, contingent on the previous tests being negative. Recent advances such as chromosomal microarray analyses and several single-gene/gene-panel DNA tests have increased the diagnostic yield. However, it is well known clinically that a diagnosis often remains elusive after this “traditional approach,” even when the index of suspicion of a genetic condition is very high.
Although the rate of unidentified genetic conditions is conventionally thought to be 50% overall, there are surprisingly few empirical data supporting this statistic. In particular, it is currently unknown how often patients with a presumed genetic disease remain undiagnosed following traditional genetic testing as defined above. Estimates have been made for individuals with specific clinical presentations such as developmental delay/cognitive disability and autism spectrum disorders; in these phenotypes, the diagnosis rate varied from 5 to 50% and as high as 80% for intellectual disabilities in a tertiary child neuropsychiatry institute.5,6,7,8,9,10,11,12,13,14 These studies were all not only specific to particular presentations but were also all published before the advent of microarrays. Subsequent studies are also limited because they focused on the diagnostic utility of chromosomal microarray or metabolic testing in determining the etiology of intellectual disabilities and/or congenital anomalies.4,15,16,17,18 In summary, the overall frequency of undiagnosed genetic disorders in a general clinical genetics setting utilizing the full range of contemporary traditional genetic testing remains unknown.
Accurate determination of the rate of unidentified genetic disorders with current diagnostic approaches is particularly important because of the increasing interest in the application of next-generation sequencing (NGS) (including whole-exome and whole-genome sequencing). With its increasing affordability, whole-exome sequencing is already being utilized in genetics clinics. Although NGS is clearly changing the diagnostic paradigm,19,20,21 it remains unclear which patients should be analyzed with NGS and at what stage in their evaluations. These questions are important to resolve in determining how best to utilize NGS and in determining which patients should continue to be evaluated with traditional genetic testing.
We completed a retrospective analysis of patients seen in the general medical genetics outpatient clinics at a tertiary medical center, to assess the diagnostic profile of an unselected consecutive cohort of patients evaluated with the current traditional approach. This included a clinical evaluation by a certified medical geneticist and diagnostic studies such as chromosomal microarray analysis, targeted single-gene and gene-panel sequencing/deletion/duplication, and biochemical and other traditional diagnostic studies. Our goals were as follows: (i) to determine the diagnosis rate, type of disorders diagnosed, number of clinic evaluations to diagnosis, and an approximation of the number/cost of the diagnostic tests performed; and (ii) to determine the appropriate patients to study with NGS and at what stage in their clinical evaluation NGS should be applied. This information provides essential data that can inform the utilization of NGS in a clinical genetics setting.
Materials and Methods
A retrospective chart review was conducted on 500 consecutive patients evaluated for diagnostic purposes between January and September 2011, in one of three general medical genetics outpatient clinics at Duke University Medical Center. Patients are referred to these outpatient clinics from North and South Carolina and Virginia; ~1,100 new and established patients are seen annually by one of three board-certified medical geneticists, with established patients typically coming back yearly for a follow-up visit.
The study was approved by the institutional review board at Duke University Medical Center. To allow us to document the number of clinic visits required to establish a diagnosis, we included all new and established patients assessed for a genetic diagnostic evaluation. We required that the patient be undiagnosed at the time of the initial genetics evaluation. Inpatients subsequently seen for a diagnostic evaluation in the outpatient clinic were considered to be established patients. Patients in the following categories were excluded from chart review: (i) those referred with a confirmed genetic diagnosis and (ii) those seen in any of the specialty genetics clinics (including metabolic, 22q11.2 deletion, Down syndrome, and fragile X syndrome clinics).
The electronic medical records were reviewed by an experienced board-certified genetic counselor and a trained research assistant, in collaboration with the three medical geneticists. Age, sex, and race of the patient, reason(s) for diagnostic referral, the patient’s medical history, and a three-generation family history were extracted. The phenotypic features on a clinical examination and the genetic laboratory investigations were recorded. If a genetic diagnosis had been made, the type of specific diagnosis was noted. If a diagnosis was either made or first considered in a clinic visit, that visit was counted as the one in which the diagnosis was established, even if confirmatory molecular testing was done subsequently, because some patients had reflexive or simultaneous studies ordered without an intervening clinic visit. We counted only the medical genetics visits and did not include other specialist visits. A separate category was created for patients who were thought not to have a genetic disorder after having been evaluated by the geneticist, with a clinical assessment and in some instances pertinent laboratory investigations.
The number and types of genetic laboratory tests were tabulated for each patient and included a karyotype, chromosomal microarray analysis, fluorescence in situ hybridization, single-gene tests, gene-panel testing, specialized genetic biochemical laboratory tests (such as urine organic acid analyses, acylcarnitine profile, and plasma amino acids), and studies for the mitochondrial genome. These tests comprised the “traditional testing.” Some of the genetic tests were ordered by other specialists before the genetic evaluation and were included.
The total costs of the genetic laboratory investigations were estimated on the basis of the tests captured from the clinic notes of the patients. We did not include in our calculation the number or costs of nongenetic laboratory investigations such as scans, radiographs, and blood chemistry. The expenses reflect the current direct/insurance (not institutional) charges for each test from the laboratories utilized most frequently by our clinicians.
A database was developed using the statistical program SPSS (version 19, IBM SPSS Statistics/9.0; IBM SPSS, Chicago, IL). The history and examination findings and the laboratory investigations were entered as quantitative variables whenever possible, to permit statistical analyses. The quantitative analyses consisted of descriptive and χ2 analyses, Fisher’s exact test, and Student’s t-test where indicated.
Results
Demographics and reasons for referral
Of the 500 patients, 281 were male (56.2%). The racial distribution included 307 Caucasians (61.4%), 115 African Americans (23%), 49 Hispanics (9.8%), 11 Asians (2.2%), and 18 others (3.6%). Other demographic details are presented in Table 1 .
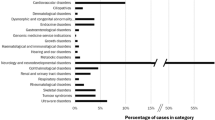
The reasons for referral were categorized, and the proportion of patients in each category were (i) developmental delay/intellectual disability (10%), (ii) congenital malformation (9.6%), (iii) family history of a definite or suspected genetic condition (4.8%), (iv) more than one reason (34.6%), (v) other specified reason (11%), (vi) suspicion of a specific diagnosis (26.2%), and (vii) referral due to a variant of unknown significance on a genetic test (3.8%). As compared with the children, adult patients had different reasons for referral, with most being referred for a suspected specific diagnosis (50 vs. 23%) and/or a positive family history (23 vs. 3%), and virtually none with malformations, developmental delay, or more than one reason.
Rate of diagnoses
After the clinical evaluation, 39 of the 500 patients were assessed as not having a genetic disorder. These included 12 patients who tested negative for a specific condition in their family. The types of laboratory investigations performed in 25 of 39 patients to aid in determining that they did not have a genetic diagnosis included those for 12 patients who tested negative for a familial mutation such as cystic fibrosis, fragile X carrier status, and familial adenomatous polyposis coli. Of the remaining 461 cases, 212 (46%) were given a genetic diagnosis based on the traditional approach. No significant differences in the rate of genetic diagnoses were observed between adults (20/38, 53%) and children (192/423, 45%) (Fisher’s exact test P = 0.65). The remaining 249 (54%) were evaluated by traditional means and believed to have a presentation consistent with a genetic disorder but remained undiagnosed.
Types of diagnoses
The 212 patients diagnosed had a variety of chromosomal and single-gene disorders, associations, sequences, teratogenic effects, and epigenetic disorders. The details are presented in Table 2 . The types of diagnoses in the adults were significantly different from those of the children (χ2 = 13.4; P < 0.05), with the adults diagnosed more frequently with single-gene disorders. The details are listed in Supplementary Table S1 online.
Number of visits and time to diagnosis
For the diagnosed patients (n = 212), the mean number of visits to reach a diagnosis was 1.52 ± 1.36, whereas those who remained undiagnosed had cumulative clinic visits of 1.88 ± 1.37 (t = 2.85; P < 0.01). The minimum and maximum visits for those with and without a genetic diagnosis were 1–16 and 1–11, respectively (the only patient to have received a diagnosis at visit 16 was detected to have a copy-number variant with the Affymetrix 6.0 microarray (Affymetrix CytoScan; Affymetrix, Santa Clara, CA) at the last visit; a previous BAC array had not shown the deletion). Of the patients receiving a genetic diagnosis, 153 (72%) were diagnosed at visit 1, and 38 (18%) patients were diagnosed at visit 2. Decreasing numbers of diagnoses were made subsequently, with only 10% (21/212) diagnosed at visit 3 or after ( Figure 1 ). The average number of years from the time of the initial evaluation to the time when a diagnosis was made was 0.63 ± 1.6 years. The undiagnosed group of patients were seen for an average of 0.99 ± 1.6 years (t = 2.36; P < 0.05). If the patient who was diagnosed at visit 16 is excluded, the mean time to diagnosis for the genetic diagnosis group would fall to 0.37 ± 1.24 years.

The majority of the 212 patients who received a genetic diagnosis with the traditional genetic paradigm did so during visit 1, with diminishing numbers of diagnoses with subsequent visits.
Number and types of genetic laboratory investigations performed
For the entire sample of 461 patients, a total of 1,314 genetic laboratory investigations were performed (see Supplementary Table S2 online). A significantly greater number of tests were performed on undiagnosed patients (3.53 ± 3.4; range: 0–33) as compared with those who were diagnosed (2.06 ± 2.21; range: 0–15; t = 5.61; P < 0.001) ( Figure 2 ). A total of 416 biochemical investigations were performed on 123 patients, and 4 were diagnostic. A total of 471 targeted single-gene tests were performed on 243 cases, with 60 leading to a diagnosis. The most commonly ordered single-gene tests in order of frequency were: fragile X, MECP2, Angelman/Prader–Willi methylation PCR, NF1, and PTEN. The details of the individual laboratory investigations are given in Supplementary Table S2 online. Of the 212 patients who received a genetic diagnosis, 87 patients (41%) were given the diagnosis using only clinical criteria—this included conditions such as neurofibromatosis type 1, for which clear diagnostic criteria exist, and others that are diagnosed using only clinical signs, such as sequences and teratogenic disorders. The other 125 patients (59%) had had confirmatory laboratory testing that clinched the diagnosis. Twenty-four patients in the unidentified genetic diagnosis category did not have laboratory investigations performed for reasons such as waiting for insurance authorization for the testing and parents deciding to defer testing.

More patients with an unidentified genetic diagnosis had multiple genetic laboratory investigations as compared with those who did receive a diagnosis, attesting to the expense associated with not having a specific disorder identified.
Costs associated with the genetic laboratory investigations
We found a significant difference in the cost of genetic tests for the undiagnosed patients as compared with the diagnosed group (t = 3.62; P < 0.001); the mean expense for laboratory tests in the undiagnosed group was $4,720.85 (ranging from $0 to $30,438.50), and for the diagnosed patients, it was $3,285.10 (ranging from $0 to $15,037.37). The cumulative costs of laboratory tests were $1,175,491.87 in the unidentified genetic diagnosis group (n = 249) and $696,441.37 in the group with a genetic diagnosis (n = 212). We also computed the laboratory expenses associated with continuing visits in the quest for a diagnosis, after visit 1. Of the 461 patients, 153 were diagnosed at visit 1, and only 59 were diagnosed during further visits. The total laboratory expenses for patients who were not diagnosed at visit 1 (n = 308) were ($1,479,848.87), with the mean cost per diagnosis for this group amounting to $25,082.16.
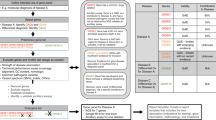
Algorithm to identify patients most suitable for NGS
On the basis of the data that were collected in this study, we developed an algorithm to guide clinicians in the identification of patients most likely to benefit from NGS ( Figure 3 ).

Algorithm to identify patients in a general genetics clinic most likely to benefit from NGS. The diagnostic process should be a clinically driven and an evidence-based interactive process, informed by the patient’s evolving clinical phenotype and the limitations inherent to laboratory investigations. As diagnostic technology advances, the testing algorithm should be reassessed. FISH, fluorescent in situ hybridization; NGS, next-generation sequencing.
Discussion
The traditional approach to diagnosing patients with suspected genetic disorders is on the threshold of a paradigm shift due to the application of NGS. Despite the justified enthusiasm surrounding NGS, critical information about when and how to best apply it remains incomplete. Our retrospective analysis of the contemporary genetic diagnostic approach identified the following salient points. Approximately 46% of patients are diagnosed with a genetic condition with the traditional approach, with the vast majority of these patients being diagnosed on the basis of the first genetics clinic visit. Of the 54% who are thought to have a genetic diagnosis but remain undiagnosed after the first visit, the average cost per subsequent genetic diagnosis is expensive, estimated minimally at $25,000. It is likely that the real cost is much greater, because we calculated only the cost of genetic laboratory tests and did not include costs of clinic visits, nongenetic laboratory tests, or procedures.
Diagnostic rates with NGS have varied in part due to differences in the types of patients from one study to another, with rates of up to 25% being reported.22,23 We recently published data demonstrating that the rate of diagnosis using NGS in general genetics clinic patients who are refractory to traditional genetic testing is 50% when specific criteria are used to optimize patient selection.19 Using the 50% diagnostic rate estimate, we can roughly approximate the cost per successful genetic diagnosis if NGS is applied after visit 1 to be $20,000 (assuming a cost of $10, 000 per patient trio for NGS on a commercial basis, because sequencing of the parents as well as the proband is currently the most effective method of identifying de novo mutations). This would still be less than the cost with continual traditional testing and if performed on a research basis would amount to one-third of the expected cost per diagnosis (assuming a cost of $4,000 per trio). Therefore, it is economically feasible to apply NGS in a clinical setting for patients who are undiagnosed after traditional testing, and this cost savings is likely to increase, with the likelihood that the costs of NGS will decrease. Therefore, we suggest that NGS should be considered if the first careful clinical evaluation and laboratory investigations do not yield a diagnosis. In addition to increasing the likelihood of a diagnosis, continued accrual of expenses due to further evaluations could be forestalled with the timely application of NGS.
We also found that adults had higher rates of diagnosis than children with the traditional approach, probably due to the fact that adults are referred often with a family history of a specific disorder or a strong suspicion of a specific condition as compared with children who are most often referred with multiple nonspecific concerns. Such children are more likely to have rare, unidentified genetic conditions and may be most amenable to NGS.
It is essential to appreciate, however, that NGS cannot be utilized as a substitute for a careful clinical evaluation, for the following reasons. Most fundamentally, we found that 6% of patients in our cohort were thought not to have a genetic condition after careful evaluation. Of note, the traditional targeted testing has a high yield. The clinician must also consider the limitations of NGS. A significant number of disorders may not be detected by NGS, such as trinucleotide-repeat disorders, mitochondrial DNA mutations, and disorders of epigenetic regulation. If such disorders have not been carefully considered before NGS, the clinician may be lulled into a false sense of security that the adaptation of NGS completes the diagnostic evaluation for the patient. Other inherent challenges with NGS include ethical controversies regarding how much/which information should be conveyed to patients and/or their families.24,25 Finally, the interpretation of the NGS data is greatly facilitated by reference to detailed clinical phenotyping.26
There is a necessity to develop clinical guidelines that would optimize patient selection for NGS. Our data and experience with NGS provide us with a framework to set forth an algorithm to facilitate the incorporation of NGS into the clinical setting ( Figure 3 ). The most critical component of this algorithm is the clinical evaluation, which should take into consideration the evolving clinical phenotype, emerging diagnostic studies, and the strengths and limitations of the tests. In the majority of patients in our cohort who received a diagnosis (category 1 in the algorithm), this diagnosis was considered and diagnostic testing was performed within the first or second clinical evaluation. For example, of the 212 patients who were diagnosed, approximately two-thirds were diagnosed on the basis of targeted laboratory tests. For patients in category 2, in which the presenting phenotype is more nonspecific or heterogeneous, if the diagnostic yield of currently available tests is low and the suspicion of an unidentified disorder is high, NGS should be utilized, after examining for copy-number variants by a chromosomal microarray. We advocate the utilization of chromosomal microarray in these instances due to the high positive rate (20%) with this test in our cohort (Table 4) and because whole-exome sequencing could miss copy-number variants. In addition, the turnaround time, complexity of interpretation, and possible detection of incidental findings27 make the option of performing a microarray before sequencing desirable. However, the continuing evolution of NGS, including the possible clinical availability of whole-genome sequencing, could result in a chromosomal microarray not being a prerequisite. Patients in category 3 may be clinically diagnosed but may not be amenable to NGS at this time, but over time at least some patients in this category may benefit from it (e.g., those with multifactorial disorders). Category 4 would include patients such as the 39 patients in our study who would be thought not to have a genetic disorder after an evaluation by a geneticist.
The limitations of our report are all inherent to the retrospective nature of the study. Clinical as well as laboratory test information was collected only from the clinic notes of the patients, and thus, we may be missing information. We did not use specific criteria to select patients who were thought not to have a genetic diagnosis, because this was not a prospective study. The genetic diagnosis rate of 46% in our study for patients with diverse clinical manifestations is likely to be spuriously low because we did not include patients followed in other specialty clinics in our medical center. Moreover, we may not have been able to accurately determine the length of time that the undiagnosed patients remained so or the number of visits they had incurred, because we were unable to include patients who might have given up on the diagnostic process and did not come back for an evaluation. In addition, because the study captured data only within a 9-month period, some undiagnosed patients would not have had a chance to come back for their annual follow-up visit. Nonetheless, our study provides data that are pertinent to other general genetics clinics within major medical centers in North America, wherein it is common practice for specialty genetics clinics to exist alongside the general genetics clinics. Although we have included data only from one medical center, the three clinics from which the data were obtained are independent of each other, and each medical geneticist was trained and certified through different programs. Because the cost of many genetic studies has decreased over time and we did not include nongenetic laboratory tests or the costs of clinical evaluations, the numbers provided here do not reflect the actual charges a patient may have incurred as part of his/her diagnostic evaluation. However, our cost estimate provides a conservative assessment of the economics of the genetic laboratory investigations performed and while valuable, stresses the need for a prospective and more complete assessment of the costs associated with genetic laboratory investigations.
In conclusion, our study, the first of its kind on a large patient cohort, contributes essential information that assesses the role of the traditional genetic diagnostic paradigm in the era of NGS. Recognizing that the field continues to evolve, our data lead us to recommend that the traditional approach be continued in the initial evaluation of a patient due to its high diagnostic yield. The detailed analyses of when in the course of patient management traditional genetic testing is obtained further suggest that NGS should be applied when the traditional approaches do not result in a diagnosis after the first visit to a genetics clinic. This work helps to identify patients most likely to benefit from NGS and suggests that its timely application would result not only in a higher rate of genetic diagnosis but a considerable cost savings per successful diagnosis.
Disclosure
The authors declared no conflict of interest.
References
Costa T, Scriver CR, Childs B . The effect of Mendelian disease on human health: a measurement. Am J Med Genet 1985;21:231–242.
Rimoin DL, Connor JM, Pyeritz RE . Nature and frequency of genetic disease, Chapter 2. In : Emery AE, Rimoin DL (eds). The Principles and Practice of Medical Genetics, 3rd edn. Churchill Livingstone: Edinburgh, NY,1997:31–34.
Kumar P, Radhakrishnan J, Chowdhary MA, Giampietro PF . Prevalence and patterns of presentation of genetic disorders in a pediatric emergency department. Mayo Clin Proc 2001;76:777–783.
Sagoo GS, Butterworth AS, Sanderson S, Shaw-Smith C, Higgins JP, Burton H . Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta-analysis of 19 studies and 13,926 subjects. Genet Med 2009;11:139–146.
Stevenson RE, Hall JG . Human malformations and related anomalies. In: Oxford Monographs on Medical Genetics, 2nd edn. Oxford University Press, 2006;3–75.
van Karnebeek CD, Scheper FY, Abeling NG, et al. Etiology of mental retardation in children referred to a tertiary care center: a prospective study. Am J Ment Retard 2005;110:253–267.
Majnemer A, Shevell MI . Diagnostic yield of the neurologic assessment of the developmentally delayed child. J Pediatr 1995;127:193–199.
Hunter AG . Outcome of the routine assessment of patients with mental retardation in a genetics clinic. Am J Med Genet 2000;90:60–68.
Challman TD, Barbaresi WJ, Katusic SK, Weaver A . The yield of the medical evaluation of children with pervasive developmental disorders. J Autism Dev Disord 2003;33:187–192.
Shahdadpuri R, Lambert D, Lynch SA . Diagnostic outcome following routine genetics clinic referral for the assessment of global developmental delay. Ir Med J 2009;102:146–148.
Schaefer GB, Lutz RE . Diagnostic yield in the clinical genetic evaluation of autism spectrum disorders. Genet Med 2006;8:549–556.
Battaglia A, Bianchini E, Carey JC . Diagnostic yield of the comprehensive assessment of developmental delay/mental retardation in an institute of child neuropsychiatry. Am J Med Genet 1999;82:60–66.
van Karnebeek CD, Jansweijer MC, Leenders AG, Offringa M, Hennekam RC . Diagnostic investigations in individuals with mental retardation: a systematic literature review of their usefulness. Eur J Hum Genet 2005;13:6–25.
Moog U . The outcome of diagnostic studies on the etiology of mental retardation: considerations on the classification of the causes. Am J Med Genet A 2005;137:228–231.
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–764.
Shen Y, Dies KA, Holm IA, et al.; Autism Consortium Clinical Genetics/DNA Diagnostics Collaboration. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics 2010;125:e727–e735.
Moeschler JB, Amato RS, Brewster T, et al. Improving genetic health care: a Northern New England pilot project addressing the genetic evaluation of the child with developmental delays or intellectual disability. Am J Med Genet C Semin Med Genet 2009;151C:241–254.
Engbers HM, Berger R, van Hasselt P, et al. Yield of additional metabolic studies in neurodevelopmental disorders. Ann Neurol 2008;64:212–217.
Need AC, Shashi V, Hitomi Y, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet 2012;49:353–361.
Rainger J, Bengani H, Campbell L, et al. Miller (Genee-Wiedemann) syndrome represents a clinically and biochemically distinct subgroup of postaxial acrofacial dysostosis associated with partial deficiency of DHODH. Hum Mol Genet 2012;21:3969–3983.
Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med 2011;13:255–262.
Gahl WA, Markello TC, Toro C, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med 2012;14:51–59.
de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921–1929.
Biesecker LG . Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: lessons from the ClinSeq project. Genet Med 2012;14:393–398.
Biesecker LG, Burke W, Kohane I, Plon SE, Zimmern R . Next-generation sequencing in the clinic: are we ready? Nat Rev Genet 2012;13:818–824.
Hennekam RC, Biesecker LG . Next-generation sequencing demands next-generation phenotyping. Hum Mutat 2012;33:884–886.
Johnston JJ, Rubinstein WS, Facio FM, et al. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet 2012;91:97–108.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(DOC 32 kb)
Supplementary Table S2
(DOC 29 kb)
Rights and permissions
About this article
Cite this article
Shashi, V., McConkie-Rosell, A., Rosell, B. et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med 16, 176–182 (2014). https://doi.org/10.1038/gim.2013.99
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.99
Keywords
This article is cited by
-
Investigating genomic medicine practice and perceptions amongst Australian non-genetics physicians to inform education and implementation
npj Genomic Medicine (2023)
-
Surgically Correctable Congenital Anomalies: Reducing Morbidity and Mortality in the First 8000 Days of Life
World Journal of Surgery (2023)
-
Real-world economic evaluation of prospective rapid whole-genome sequencing compared to a matched retrospective cohort of critically ill pediatric patients in the United States
The Pharmacogenomics Journal (2022)
-
Birth defect co-occurrence patterns in the Texas Birth Defects Registry
Pediatric Research (2022)
-
Toward the diagnosis of rare childhood genetic diseases: what do parents value most?
European Journal of Human Genetics (2021)
