
Abstract
Population genetic studies on European populations have highlighted Italy as one of genetically most diverse regions. This is possibly due to the country’s complex demographic history and large variability in terrain throughout the territory. This is the reason why Italy is enriched for population isolates, Sardinia being the best-known example. As the population isolates have a great potential in disease-causing genetic variants identification, we aimed to genetically characterize a region from northeastern Italy, which is known for isolated communities. Total of 1310 samples, collected from six geographically isolated villages, were genotyped at >145 000 single-nucleotide polymorphism positions. Newly genotyped data were analyzed jointly with the available genome-wide data sets of individuals of European descent, including several population isolates. Despite the linguistic differences and geographical isolation the village populations still show the greatest genetic similarity to other Italian samples. The genetic isolation and small effective population size of the village populations is manifested by higher levels of genomic homozygosity and elevated linkage disequilibrium. These estimates become even more striking when the detected substructure is taken into account. The observed level of genetic isolation in Friuli-Venezia Giulia region is more extreme according to several measures of isolation compared with Sardinians, French Basques and northern Finns, thus proving the status of an isolate.
Similar content being viewed by others


Introduction
Human complex traits arise from the new mutations as well as from the interplay between existing genetic variants and exposure to environmental conditions. This means that it is desirable to study populations with decreased genetic variability, such as isolated populations1, 2 and large homogeneous populations2, 3 as more power is gained for genetic association mapping studies.4 Population isolates are by definition characterized by small effective population size (Ne), which results in stronger effects of random genetic drift leading to decreased genetic variability.5 These processes can be temporally continuous and/or woven into a chain of discrete bottleneck events in the demographic history of an isolate. Several population isolates, like Sardinians,6 northern Finns,4 Amish7 and Icelanders8 have successfully been used in both linkage and genome-wide association studies for pinpointing DNA sequence variants for disease predispositions (OMIM database: www.omim.org and NHGRI GWAS Catalog9). Another favorable aspect of population isolates is the largely shared environment. The isolation usually arises from geographical barriers and therefore everyone is exposed to the same factors, which enables to effectively design powerful gene–environment interaction studies.5
In recent years, several studies have started to shed light on the structure of the genetic variation on global,10, 11 continental,12, 13 regional,3, 14 ethnic group (such as Jews,15 Indians,16 Brazilia17) and country2, 3, 18, 19 level, but very rarely are the studied sample representative for a whole country.2, 3 The analyses on the sub-population level20, 21 focus mostly on population isolates and demonstrate a decreased genetic variability within, but also elevated diversity between neighboring regions and source populations.2, 15, 21, 22 It is important to characterize any putative population isolate for events of recent admixture and presence of sub-structure as these could disrupt the genetic homogeneity and lead to possible spurious associations in gene mapping studies.23 Therefore, a correct sampling strategy is fundamental.
Italians, on the example of Sardinians, are one of the most studied European populations next to Saami people and Basques where the population structure has been analyzed in depth by using both the haploid loci (mtDNA and Y chromosome)24, 25 and the autosomal markers.3, 20 It is hypothesized that Italy may be enriched for population isolates because of complex demographic history and topographic variability (Italian Network of Genetic Isolates: http://www.netgene.it/ita/ingi.asp).
This study aims to test this hypothesis by genetically characterizing a hilly part of Friuli-Venezia Giulia (FVG) county located in northeastern Italy. The region is particularly interesting, – while covering a total area of only 7858 km2 several distinct dialects are spoken and several villages sport traditions and/or surnames linking them to ethnic groups further away rather than in FVG or in Italy for that matter. All in all, it seems the area is characterized by complex demographic history. For example, people in the village of Resia speak an archaic proto-Slavic language, known as Resian, but their surnames are Italian or Italianized. In the village of Illegio, not far from Resia, people speak another local language – called friuliano – of the Rhaeto-Romance language sub-family, which, during the Middle Ages, was widespread – from modern Switzerland to Slovenia. Illego is further characterized by a limited number of surnames, what could be interpreted as evidence for marginal immigration. Another layer of specific cultural heritage is added by characteristic local symbols found engraved on local houses. The inhabitants of village Sauris speak an archaic dialect of German origin and according to legends the locals have their ancestral roots near Tyrol. Until 50 years ago, before a flood devastated the valley of origin, the main spoken language in the village of Erto was a Latin dialect called ertano, while current population is an admixture of the former inhabitants of Erto and migrants from the nearby regions. The village of San Martino del Carso (SMC) is the only Italian-speaking village in the Slovenian-speaking Carso region. Finally, the village of Clauzetto is located in a remote valley where people speak friulano.
The rich history and linguistic diversity of these six populations allows to predict elevated levels of intragroup genetic homogeneity, higher intergroup differentiation between the villages, and suggests at least some degree of ancestry with the neighboring regions of Slovenia, Germany and Italy. In this study, we analyze the genetic variation within the FVG region in order to understand (a) the relationship between the villages inhabitants and the other Italians, and Europeans as a whole, which may offer unique insight into the complex demographic history of the region, and (b) to evaluate if any of the village populations represent a genuine population isolate, as these would have a great potential in genetic epidemiological studies. In order to answer these questions, high-density genotype data of >1400 newly analyzed samples from FVG region and Slovenia were combined with several publicly available data collections.3, 11, 15, 16, 26 This enables us to directly contrast the hypothetical cultural origins of FVG region villages against the data-driven genetic similarity patterns. Furthermore, we compare the genetic diversity and genomic homogeneity found in the six FVG villages with other well-known geographical and cultural population isolates, such as Sardinians, French Basques and northern Finns.
Materials and methods
Samples
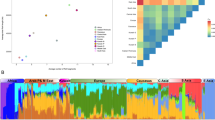
We genotyped 1310 samples from six geographically isolated villages in the Italian FVG region. We combined this data with the 96 newly genotyped Slovenian samples and with five published population-based collections3, 11, 15, 16, 26 (see Supplementary Table 1), with an emphasis on populations with European ancestry. After data quality control and exclusion of close relatives, 3091 samples from 72 populations were used for the analyses (Supplementary Table 1). This set included 733 samples of Italian ancestry from 11 populations: (1) Borbera Valley (North-West Italy) and Apulia region (South-East Italy; Carlantino)3 and (2) Sardinians, Tuscans and northern Italians from Human Genetic Diversity Panel11 and (3) six villages from the FVG region. Figures 1a and b shows the valleys and isolated villages from where the Italian samples were collected. Hereafter, the nation name is used for general populations and a village or region name for more isolated populations.

Map showing approximate location of analyzed Italian samples and revealed population structure in analyzed populations. (a) A geographical map of Italy and shown are the approximate sampling regions. (b) A detailed geographical map of FVG and shown are the approximate location of the six villages. (c) Ancestry proportions of the studied 1008 individuals from 39 European and Near-Eastern populations (including the six FVG village populations) as revealed by the ADMIXTURE program29 with K=2 to K=10. A stacked column of the K proportions represents each individual, with fractions indicated on the y axis. From all non-FVG populations a subset of 24 randomly drawn individuals (if applicable) was used.
A written informed consent for participation was obtained from all newly genotyped subjects.
Single-nucleotide polymorphism (SNP) genotypes and quality control
The six Italian cohorts were genotyped using the Human370CNV and the Slovenian sample with the HumanOmniExpress beadchips according to the manufacturer’s protocol (Illumina Inc., San Diego, CA, USA). The published data sets used in the analysis had been genotyped with different versions of Illumina beadchips.
The genotype data from different platforms were first merged and then filtered according to the standard genotype quality control metrics using PLINK v1.07 software.27 Only the SNPs of the 22 autosomal chromosomes with minor allele frequency of >1%, Hardy–Weinberg equilibrium P-value >10-6, and genotyping success rate >95% were included, leaving approximately 145 000 SNP markers. Only the individuals with genotyping success rate >95% were used. Cryptic relatedness was tested with the same software and from the detected relative pairs (up to the second cousins) only one was randomly chosen for the subsequent analyses. Depending on the analyses (eg, computational optimization and bias in sample sizes) very distant ethnic groups were excluded and the population sample sizes were trimmed to 50 or 24 samples. Background linkage disequilibrium (LD) can induce bias in the principal component28 and structure-like analyses; therefore the set of 145 000 SNPs was thinned by excluding markers in strong LD (pairwise genotype correlation r2>0.4) in a window of 200 SNPs (sliding window overlap 25 SNPs at a time) leaving approximately 101 000 SNPs for the subsequent analyses.
Statistical analyses
An unsupervised, maximum likelihood-based clustering algorithm assembled in ADMIXTURE29 software was applied to the European and Near-Eastern ancestry (Jews, Palestinians, Adyghes and Druzes) population samples (n=1975) to identify the putative ancestral clusters within the samples as well as to assess the extent of admixture. Clustering was performed 100 times at K=2 to K=15 and the best-fitting K was selected according to the lowest cross-validation (CV) index (Supplementary Figure 1A). In addition, we opted to use a threshold level of variation in log likelihood scores (LL<1) within a fraction (10%) of runs with the highest LLs15 (Supplementary Figure 1B) as a pointer to assume that the global likelihood maximum was reached, thus rendering the given K model a useful representation of the genetic structure of the sample. The lowest CV indexes were observed at K=9 while K=10 showed only marginally worse values. It is likely that global likelihood maximum was indeed reached at K=2 to K=15.
For several analyses, the FVG populations were split into sub-populations according to ancestry estimations at K=10 (if applicable) as follows: (1) general set (GS), when village-specific ancestry loading was smaller than 30% and (2) more isolated set (IS), when loading exceeded 30%. We choose K=10 to discern between the sets because beyond this K (and up to K=15, see above) no additional village-specific components arose, thus making this K the most appropriate one to choose for this particular task. This choice was supported by observing that for K=10, (1) the global likelihood maximum was indeed probably reached and (2) the CV index values were close to the lowest ones at K=9. Subsequently, a sub-set of 24 individuals was chosen at random from each European and Near-Eastern ancestry population for the following analyses in order to minimize sample size effects.30 In all instances, the sampling was repeated five times and the obtained pairwise Fst distances were close to unity, indicating that the random sets were representative of the entire sample.
Principal component analysis (PCA) using 101 000 SNPs was performed using EIGENSOFT31 package. The same software was used to calculate the pairwise Fst values that were further analyzed with hierarchical agglomerative clustering. The analyzes were performed on two sets of samples: (1) all available samples (n=3091), and (2) only the trimmed European and Near-Eastern ancestry populations (n=1008). Spatial ancestry analysis (SPA) was applied on the latter set, to explicitly model the spatial distribution of each marker in order to describe the degree of population stratification in each of the putative population isolates into the European genetic background.32
Principal component ancestry informative markers (PCAIMs)33 panel was constructed of 250 SNPs (out of 101 000 SNPs) retained from the top five PCs (50 SNPs from each)17 ranked by the absolute loading scores. PCAIMs panel was the input to a non-model-based multivariate approach, a discriminant analysis of principal components (DAPCs), which is implemented into the R package adegenet ver1.3-0.36 All principal components were included to the k-means clustering algorithm from KDAPC=1 to KDAPC=14, and the best-fitting KDAPC was selected using the Bayesian information criterion. DAPC was used to assign individuals into the predicted clusters and real populations were used as identifiers. Over fitting was avoided by estimating the difference between the proportion of successful reassignments and values obtained using random grouping.
Pairwise LD extent was calculated as the genotype correlation (r2) between marker pairs <100 kb apart using the PLINK v1.0727 software. A custom Perl script was applied to categorize the r2 values according to inter-marker distances (0−5 kb, 5−10 kb, and so on) and mean r2 was calculated for each category. Calculations were restricted only to the established population isolates, the FVG region samples, and a set of geographical reference populations (Estonians, Slovenians and Swiss).
The genomic runs of homozygosity (gROH) and the inbreeding coefficient (Fin) were estimated using PLINK v1.07 with established parameters.34 ROH was defined as a sequence of at least 25 consecutive homozygous SNPs spanning at least 1500 kb, with a maximum gap of 100 kb between the adjacent SNPs and a density of SNPs within the run of no >20 kb/SNP34 (for each individual the gROH was defined as the sum of above defined genomic regions). The Fin was estimated for each sample based on the ratio between observed and expected number of the homozygous genotypes.
Results
FVG region in the context of broader European genetic diversity
As the first step in understanding the place of the FVG region populations on the genetic canvas of Europe, we applied a model-based structure-like algorithm assembled in ADMIXTURE29 to compute quantitative estimates for individual ancestry in K hypothetical ancestral populations. The best predictive accuracy was observed for a model with K=9 (Supplementary Figure 1). In general, the ancestry proportions distributions among FVG village samples were very similar to other populations in the geographical region, although on higher K values almost all of the FVG populations became dominated by a single component largely specific to any one particular village. The results revealed a substantial level of intra-population structure in most of the FVG region populations manifested in elevated variability in membership to the village-specific ancestry component (Figure 1c). It is likely that this structure is representing the intra-population differences in level of inbreeding rather than specific ancestry. This interpretation gains further support in subsequent analyses where the presence of the village-specific ancestry component was found to be highly correlated with both extended LD range and elevated levels of gROH (Supplementary Figure 7; see below). For further analyses, the village populations were split into sub-populations (referred as GS and IS) based on the prevalence of the village-specific ancestry component (described in Materials and Methods section) and considered as independent populations to more accurately represent the genetic diversity within FVG villages.
Next, we conducted a PCA on the European and Near-Eastern ancestry populations (on the global PCA plot11 all FVG samples clustered onto Europeans; Supplementary Figure 2A). The two first principal components accounted for 1.5% and 0.7% of the genetic variation, whereas clearly separating the population isolates, like Sardinians,22 French Basques22 and northern Finns,2 but also the more homogenous FVG sub-populations (Figure 1; Supplementary Figure 2B). A more detailed picture emerged from the SPA32 analyzes as follows: (a) SMC has the widest spread, clustering onto the German-speaking populations and partially overlapping with the Slovenians, (b) the GS sub-populations of Clauzetto, Sauris and Illegio cluster between SMC and northern Italians, from which Borbera and Tuscans positioned farther ‘south’ and (c) GS sub-populations of Erto and Resia are shifted toward population isolates but cluster less distantly (Figure 2). The clustering of the FVG region samples in both PCA and SPA projections were roughly representative to their geographical location.

Model-based mapping convergence with SPA. Label position indicates the (a) specific PC1 and PC2 coordinate values for each individual and (b) the mean PC1 and PC2 coordinate values for each population. For (a, b), the colors have a following meaning: (1) dark blue color: a homogeneous fraction of the FVG population; a blue color: more general fraction of the FVG population; a red color: other Italian samples; a violet color: Basques; an orange color: Slovenians; and green color: all other populations. For (a, b), the following population abbreviation labels are used: AT, Austrians; BA, French Basques; BG, Bulgarians; BO, Borbera; CA, Carlantino; CL, Clauzetto; CH, Swiss; CZ, Czechs; GR, Germans; ER, Erto; ES, Spaniards; FR, French; HU, Hungarians; IL, Illegio; IT, Italians; JW_A, Ashkenazy Jews; JW_S, Sephardic Jews; OR, Orcadians; RE, Resia; RO, Romanians; SA, Sardinians; SA_, Sauris; SMC, San Martino del Carso; SI, Slovenians; TU, Tuscans. The extra abbreviations: N, northern; S, southern; I, a more homogeneous sub-population; G, a more general sub-population.
To further minimize the bias from within- and maximize the between-group variance, we applied the DAPCs35 to more precisely identify the genetically closest populations to the FVG groups. For the combined European and Near-Eastern populations, the SNP PCAIM panel had the best fit for KDAPC=7 (Supplementary Figure 3) and the DAPC clearly out-clusters the IS sub-populations from Illegio (C1), Sauris (C3) and Resia (C6), including some of the respective GS sub-population samples as well (Figure 3). The rest of the FVG populations clustered mostly with C2 (dominated by central Europeans) and C5 (dominated manly by Italians), although a minor fraction was also assigned to C4 (dominated by northern Europeans), like Sauris and SMC, and C7 (dominated by Near-Eastern ancestry populations), like Clauzetto, Illegio and Sauris but also other southern Europeans (Figure 3).

Predicted cluster membership for each individual from DAPCs. Model optimum was found on K=7 and individuals are assigned to best-fitting cluster. The size of the rectangle scales with the number of assigned samples. In population names: N, northern; S, southern; I, a more homogeneous sub-population; G, a more general sub-population.
Hierarchical agglomerative clustering analysis based on the between populations pair-wise Fst distances revealed that the IS sub-populations were very distant from all other populations (Supplementary Figure 4). The GS sub-populations showed very small pair-wise Fst distances from the geographically close populations (Supplementary Figure 5). Two main observations were made: (1) the SMC was equally distant from Sauris and Clauzetto as well as from its geographical neighbors, such as Slovenians, and (2) Sauris resembled Clauzetto the most but also the Swiss. All the other FVG sets resembled a larger list of populations (Supplementary Figure 5).
FVG village populations as genetic isolates
Some of the FVG villages reside in very remote valleys and this can lead to genetic isolation, so we next investigated measures of genetic diversity that might reflect a history of relative isolation, elevated levels of inbreeding and/or recent historical bottlenecks. To compare the depth of the isolation in the FVG village sub-populations, we compared the observed values with the established population isolates (like French Basques, Sardinians and northern Finns) but also with the other reference populations.
A high level of inner-structure in the FVG populations was revealed by ADMIXTURE (Figure 1c), which was further confirmed by the PCA, SPA and DAPC (Supplementary Figure 2B; Figures 2 and 3). For example, in both the PCA and SPA analyses, the IS FVG sub-populations (except Clauzetto) positioned very distantly from their geographical neighbors (Supplementary Figure 2B). Genetic isolation of FVG populations was further highlighted by DAPC, which assigned three of the IS sub-populations (Illegio, Sauris and Resia) exclusively to respective single village-specific clusters, whereas all other FVG populations were distributed between several clusters (Figure 3).
As may be expected from the observed genetic structure, the between populations pair-wise Fst values of the IS sub-populations were extremely high compared with all the other populations (overall mean Fst: Resia 0.023 and Sauris 0.021 vs SMC 0.006) and elevated compared with the previously characterized population isolates (overall mean Fst: Sardinia 0.014; French Basques 0.013 and northern Finns 0.18) (Supplementary Figure 6).
The results from the LD analysis revealed a striking difference between the sub-populations drawn from a single village compared with the established population isolates, whereas even the (GS) sub-population of Resia and Illegio showed a strong deviation in LD from the expected southern decline3 (Figure 4). The LD block length was the shortest for the GS sub-populations of Clauzetto and Sauris, where the extent was comparable with values observed for Slovenians and Swiss.

Genome-wide LD length based on 145 000 SNPs. Each line represents the LD decay averaged across populations and sub-populations and LD (r2) between SNPs shown in 5 kb bins. Sub-populations drawn from same population have the same color coding. A dashed line represents the more general sub-populations and the solid line with rectangles represents the more homogeneous sub-populations respectively. A solid thin line represents the European reference populations (Estonians, Slovenians and Swiss). The LD extent in reference population isolates (Sardinians, French Basques and northern Finns) is also shown with a solid line with rectangles. In population names: I, a more homogeneous sub-population; G, a more general sub-population.
Finally, similar large deviation from the expected values was observed for population mean gROH in IS sub-populations (Figure 5). For example, the mean gROH for the latter set was 47 Mb, compared with 11 Mb in the population isolates cluster, 5.5 Mb in the GS sub-populations and roughly 2 Mb in the European reference populations (Figure 5). The latter is similar to previously published estimates.36, 37 The same tendency was observed from clumped inbreeding coefficient estimates respectively in above defined clusters: 0.04, 0.028, 0.009 and 0.011 (Supplementary Table 2). These two estimates were highly correlated with the village-specific ancestral components (Supplementary Figure 7) and thus it is probably the decreased diversity because of small Ne in the subsets of villages that is behind the striking structure.

The genomic runs of homozygosity (gROH) based on 145 000 SNPs. (a) Distribution of population mean homozygous segments and mean count of gROH>1.5 Mb per sample. (b) Zoom in view for the region indicated with a dashed ellipse on (a). FVG sub-populations are indicated in bold letters. In population names: I, a more homogeneous sub-population; G, a more general sub-population.
Discussion
In this study, we were interested to see how the FVG village populations fit into the general paradigm of spatial structure of the genetic variation of European populations using 145 000 autosomal SNPs. To this end, we compared the genetic variation of the FVG village populations against a reference panel composed of other Italian, a wider range of European, and a set of Near-Eastern populations. We observed a striking level of genetic structure among the FVG populations (Figure 1c), which likely arises from increased genetic similarity within specific subsets of samples from respective villages and not difference in genetic origin. The FVG-specific components were present in the background profile of all Europeans, which is consistent with the FVG-specific components representing a fraction of the overall genetic variability in Europe.
Based on the composition of ancestral components the FVG populations were split into two sets for the subsequent analyses: (1) more general sub-population (GS), which resembled other Italians and populations in close proximity, and (2) more diverse and very homogeneous set (IS). The GS sets were used to further understand the genetic position of the FVG populations on the European genetic canvas and to look for the genetic roots of cultural differences. The two first components in both the PCA and the SPA analyses positioned the FVG samples close within the variation of Italian populations as reported by Li et al.11 In addition, FVG region showed the smallest Fst distances with the populations in close geographical proximity. For example the SMC, while being an only Italian-speaking village in a Slovenian-speaking region, had equally distant Fst values from Slovenians and other FVG populations, but at the same time had a very broad clustering in the SPA and was consistent with varied level of admixture. This suggests that the original Italian founders of the village have genetically mixed with the neighboring Slovenians and thus on PC plot position in between Italians and Slovenians but have maintained their Italian cultural heritage.
From the discriminant analyses of principal components, which effectively minimized the large within-group variability,33 we found some hints for the origin of Sauris, the historical founders of which are believed to be German speakers from the region adjacent to Tyrol (Supplementary Note). In the DAPC, the Sauris samples positioned to several clusters, mainly Italian-specific C5 and southern European-specific C2 but also in the northern European cluster C4 (also slightly present only for SMC), together with the Germans and Swiss. The smallest Fst distance with the Swiss next to Clauzetto further strengthened the Sauris link with the German-speaking populations.
In addition to being intriguing subjects in studies on population history, linguistics and ethnogenesis, population isolates are useful tools for genetic epidemiology studies in quest for finding disease susceptibility alleles. Using isolates is advantageous because of very similar exposure to environmental factors and effects of small Ne – enrichment of some harmful sequence variants because of more pronounced random genetic drift, a higher level of consanguinity and overall genetic homogeneity.5, 6 In this study, we detected a strong signal for genetic isolation for subsets of the village populations that could only be discovered by analyzing a representative fraction of the village populations. The more homogeneous FVG sub-populations showed more extreme values for all considered measures of isolation compared with Sardinia,11, 36 French Basques11, 36 and northern Finns.2 Most of the extensive genetic homogeneity is explained by the very small Ne within the FVG villages (eg, 105 inhabitants in the village of Erto), which has led to high but varying levels of consanguinity suggested also by the trice as large inbreeding coefficients in GS compared with IS sub-population clusters. Not surprisingly also the more general sub-populations of the FVG, except Clauzetto, demonstrated values in the direction that is indicative of isolation relative to the European reference set for Fst distances,3 in the extent of LD3 and levels of genomic background homozygosity.34, 36, 37
This study has yielded two main results. We have deeply characterized and confirmed the status of genetic isolate for four further populations, which provide a useful tool for gene mapping studies. Second, the genetic structure analyses clearly highlights the need to analyze a large and representative sample to precisely estimate the intragroup variability in a population. Our results show the extent of the genetic diversity and variation within and between populations sampled from northeastern part of Italy while highlighting an extreme level of isolation compared with other genetic isolates.
References
Price A, Helgason A, Palsson S et al: The impact of divergence time on the nature of population structure: an example from Iceland. PLoS Genet 2009; 5: e1000505.
Jakkula E, Rehnström K, Varilo T et al: The genome-wide patterns of variation expose significant substructure in a founder population. Am J Hum Genet 2008; 83: 787–794.
Nelis M, Esko T, Mägi R et al: Genetic structure of Europeans: a view from the North–East. PLoS One 2009; 4: e5472.
Peltonen L, Jalanko A, Varilo T : Molecular genetics of the Finnish disease heritage. Hum Mol Genet 1999; 8: 1913–1923.
Kristiansson K, Naukkarinen J, Peltonen L : Isolated populations and complex disease gene identification. Genome Biol 2008; 9: 109.
Orrù S, Thomas G, Loizedda A, Cox DW, Contu L : 24 bp deletion and Ala1278 to Val mutation of the ATP7B gene in a Sardinian family with Wilson disease. Hum Mutat 1997; 10: 84–85.
Puffenberger EG : Genetic heritage of the Old Order Mennonites of southeastern Pennsylvania. Am J Med Genet C Semin Med Genet 2003; 121C: 18–31.
Holm H, Gudbjartsson DF, Sulem P et al: A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet 2011; 43: 316–320.
Hindorff LA, Sethupathy P, Junkins HA et al: Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009; 106: 9362–9367.
Jakobsson M, Scholz SW, Scheet P et al: Genotype, haplotype and copy-number variation in worldwide human populations. Nature 2008; 451: 998–1003.
Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104.
Heath SC, Gut IG, Brennan P et al: Investigation of the fine structure of European populations with applications to disease association studies. Eur J Hum Genet 2008; 16: 1413–1429.
Novembre J, Johnson T, Bryc K et al: Genes mirror geography within Europe. Nature 2008; 456: 98–101.
Lao O, Lu TT, Nothnagel M et al: Correlation between genetic and geographic structure in Europe. Curr Biol 2008; 18: 1241–1248.
Behar DM, Yunusbayev B, Metspalu M et al: The genome-wide structure of the Jewish people. Nature 2010; 466: 238–242.
Metspalu M, Romero IG, Yunusbayev B et al: Shared and unique components of human population structure and genome-wide signals of positive selection in South Asia. Am J Hum Genet 2011; 89: 731–744.
Giolo SR, Soler JM, Greenway SC et al: Brazilian urban population genetic structure reveals a high degree of admixture. Eur J Hum Genet 2012; 20: 111–116.
Humphreys K, Grankvist A, Leu M et al: The genetic structure of the Swedish population. PLoS One 2011; 6: e22547.
O’Dushlaine CT, Morris D, Moskvina V et al: Population structure and genome-wide patterns of variation in Ireland and Britain. Eur J Hum Genet 2010; 18: 1248–1254.
Pistis G, Piras I, Pirastu N et al: High differentiation among eight villages in a secluded area of Sardinia revealed by genome-wide high density SNPs analysis. PLoS One 2009; 4: e4654.
Huyghe JR, Fransen E, Hannula S et al: A genome-wide analysis of population structure in the Finnish Saami with implications for genetic association studies. Eur J Hum Genet 2011; 19: 347–352.
Veeramah KR, Tönjes A, Kovacs P et al: Genetic variation in the Sorbs of eastern Germany in the context of broader European genetic diversity. Eur J Hum Genet 2011; 19: 995–1001.
Marchini J, Cardon LR, Phillips MS, Donnelly P : The effects of human population structure on large genetic association studies. Nat Genet 2004; 36: 512–517.
Simoni L, Calafell F, Pettener D, Bertranpetit J, Barbujani G : Geographic patterns of mtDNA diversity in Europe. Am J Hum Genet 2000; 66: 262–278.
Calò CM, Corrias L, Vona G, Bachis V, Robledo R : Sampling strategies in a linguistic isolate: results from mtDNA analysis. Am J Hum Biol 2012; 24: 192–194.
Yunusbayev B, Metspalu M, Järve M et al: The Caucasus as an asymmetric semipermeable barrier to ancient human migrations. Mol Biol Evol 2012; 29: 359–365.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Huma Genet 2007; 81: 559–575.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Alexander DH, Novembre J, Lange K : Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19: 1655–1664.
McVean G : A genealogical interpretation of principal components analysis. PLoS Genet 2009; 5: e1000686.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D : Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
Yang WY, Novembre J, Eskin E, Halperin E : A model-based approach for analysis of spatial structure in genetic data. Nat Genet 2012; 44: 725–731.
Drineas P, Lewis J, Paschou P : Inferring geographic coordinates of origin for Europeans using small panels of ancestry informative markers. PLoS One 2010; 5: e11892.
McQuillan R, Leutenegger AL, Abdel-Rahman R et al: Runs of homozygosity in European populations. Am J Hum Genet 2008; 83: 359–372.
Jombart T, Devillard S, Balloux F : Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 2010; 11: 94.
Kirin M, McQuillan R, Franklin CS et al: Genomic runs of homozygosity record population history and consanguinity. PLoS One 2010; 5: e13996.
Nothnagel M, Lu TT, Kayser M, Krawczak M : Genomic and geographic distribution of SNP defined runs of homozygosity in Europeans. Hum Mol Genet 2010; 19: 2927–2935.
Acknowledgements
EGCUT received financing from FP7 grants (ENGAGE 201413, OPENGENE 245536), Estonian Basic Research grant SF0180142s08, Estonian Research Roadmap through Estonian Ministry of Education and Research (3.2.0304.11-0312), Center of Excellence in Genomics (EXCEGEN) and Development Fund of University of Tartu (SP1GVARENG). M Metspalu was supported by Estonian Basic Research grant SF0270177As08 and Estonian Science Foundation grant (8973). We acknowledge EGCUT personnel, especially Mr V Soo. Data analyzes were carried out in part in the High Performance Computing Center of University of Tartu. M Macek was supported by CZ.2.16/3.1.00/24022.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Esko, T., Mezzavilla, M., Nelis, M. et al. Genetic characterization of northeastern Italian population isolates in the context of broader European genetic diversity. Eur J Hum Genet 21, 659–665 (2013). https://doi.org/10.1038/ejhg.2012.229
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.229
Keywords
This article is cited by
-
Characterization of Danube Swabian population samples on a high-resolution genome-wide basis
BMC Genomics (2023)
-
Genetic history of Calabrian Greeks reveals ancient events and long term isolation in the Aspromonte area of Southern Italy
Scientific Reports (2021)
-
The genetic structure of Norway
European Journal of Human Genetics (2021)
-
Natural human knockouts and Mendelian disorders: deep phenotyping in Italian isolates
European Journal of Human Genetics (2021)
-
Whole-genome sequencing reveals new insights into age-related hearing loss: cumulative effects, pleiotropy and the role of selection
European Journal of Human Genetics (2018)

{kind=link}
{kind=link}