
Abstract
Multiple previous reports confirm that several missense alleles of MSX1 exhibit Mendelian inheritance of an oligodontia phenotype (agenesis of more than six secondary teeth besides third molars). However, the extent to which missense MSX1 alleles contribute to common, multifactorial disorders is less certain. It is still not yet clear whether multiple non-synonomous MSX1-coding variants identified among patients with oral clefting are merely neutral polymorphisms or whether any of these might represent real mutations with mild effects. The present work steps toward resolving these issues for at least one MSX1 allele: R151S, previously identified in a single Japanese proband with unilateral cleft lip and palate. Candidate gene sequencing within a patient cohort demonstrating mild tooth agenesis (loss of six or less secondary teeth besides third molars, hypodontia), secondarily identified this same MSX1 variant, functioning as a mildly deleterious, moderately penetrant allele. Four of five heterozygous R151S individuals from one Japanese family exhibited the hypodontia phenotype. The in vitro functional assays of the variant protein display partial repression activity with normal nuclear localization. These data establish that the MSX1-R151S allele is a low-frequency, mildly deleterious allele for familial hypodontia that alone is insufficient to cause oral facial clefting. Yet, as this work also establishes its hypomorphic nature, it suggests that it may in fact contribute to the likelihood of common birth disorder phenotypes, such as partial tooth agenesis and oral facial clefting. Nevertheless, the exact mechanism in which differential pleiotropy is manifested will need further and deeper clinical and functional analyses.
Similar content being viewed by others

Introduction
Work on model systems has demonstrated that the muscle segment homeobox 1 (MSX1) locus exerts pleiotropic effects on multiple tissues, including dental and oral–facial structures. This results from the well-documented early expression domains within these tissues during embryogenesis,1 and is reflected in the phenotype of the Msx1 knockout mouse: cleft palate, tooth agenesis and nail dysplasia.2, 3 In humans, multiple MSX1 mutations have been identified in families exhibiting Mendelian inheritance of an oligodontia phenotype (either non-syndromic or syndromic).3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 However, MSX1 also clearly affects the common human oral cleft phenotypes, as demonstrated through multiple genetic studies.15, 16, 17, 18, 19, 20 Yet, in all but a single case report,5 MSX1 apparently functions as a complex disease determinant for oral clefting. In this context, there have been multiple reports of MSX1 missense mutations within sporadic clefting cases,17, 18, 21, 22 but all these reports suffer from a lack of power to discern disease risk. This is a problem that is inherent to the study of sporadic or rare variants. In an attempt to ameliorate this issue, deep phylogenomic analyses of the MSX family protein sequences helped classify and prioritize the study of the MSX1 missense variants.1 Yet it is still really not clear as to which variant and to what extent these variants might contribute to the clefting phenotype, nor how the clefting and the tooth loss phenotypes due to MSX1 mutations may be related in human populations. Further complicating the clarification of the differential pleiotropy issue is the fact that for most of the clefting studies, minor anomalies such as congenital tooth loss or nail dysplasia have historically been under ascertained. As already detailed elsewhere, it will be necessary to perform further and deeper clinical and functional analyses to properly determine these relationships.1, 23
To describe dental anomalies, several different terms are used. The most extreme situation is anodontia, denoting absence of all teeth.24 Hypodontia is defined as an absence of one to six secondary teeth, excluding third molars,25 whereas the absence of more than six teeth, again excluding third molars, is referred to as oligodontia. Approximately 20% of the human population is missing at least one secondary tooth (mostly third molars). The overall prevalence of missing other teeth (besides third molars) in humans is 1–10%, with 70–80% of cases missing either one or two teeth.6, 26 Such secondary or permanent tooth agenesis is one of the most common developmental anomalies in humans (OMIM: 106600, 604625)6, 27 and may occur independently or as one feature of a syndrome. The non-syndromic forms may be sporadic or familial, with prevalence varying by tooth type.28 This study targets familial hypodontia, a relatively mild phenotype within the tooth agenesis spectrum; that is, loss of more than just third molars but less than oligodontia or anodontia.
As mentioned above, mutations not only within MSX1 but also within Paired box homeobox 9 (PAX9) are known from patients with either non-syndromic or syndromic oligodontia.29, 30, 31, 32 Although the MSX proteins generally function as transcriptional repressors, these effects may be modulated by protein–protein interactions. Notably, the Msx1 protein has seven ancient, highly conserved sequence motifs, named Msx homology (MH) domains,1 which likely modulate its interactions with several other binding partners, including Pax9.1, 33 These two proteins also interact genetically to enhance the activation of Bone morphogenetic protein 4 expression during tooth and craniofacial development.34 The MSX1 and PAX9 transcription factors apparently operate within shared critical pathways during murine embryogenesis. This is exemplified by the common phenotypes of homozygous Msx1 or Pax9 gene knockout mice: cleft secondary palate, facial abnormalities and complete anodontia, with concomitant arrest of tooth development at the bud stage.2, 35 An allelic series of Pax9 mutants demonstrated the graded effects of dose on mouse tooth development.36, 37 Msx1 effects are also known to be dose sensitive.38, 39 Thus, there is ample evidence to suggest that MSX1 and PAX9 are excellent candidate genes for human familial hypodontia. It is also plausible that a partial dose reduction or partial functional deficit of either PAX9 or MSX1 may contribute to partial tooth agenesis.
In light of the moderate penetrance and mild tooth agenesis phenotype in a family carrying the R151S variant described below, it became necessary to examine the influence of other known, likely complex disease alleles and environmental risk factors on these phenotypes. Several previous candidate gene association reports link more common, but less penetrant, genetic variation at the MSX1 locus with tooth agenesis and/or with the oral facial clefting phenotype.15, 40, 41, 42 It is the most common fourth allele of the intronic CA repeat (CA4) that is associated with disease,43, 44 whereas the CA2 allele was significantly undertransmitted in a biallelic model of oral facial clefting,17 and may be protective for that phenotype. Other reports suggest that the tooth phenotypes may be a sub-phenotype of oral–facial clefting disorders, suggesting that the tooth and face share developmental pathway elements.45 It is conceivable that the complex disease alleles marked by these CA repeats could potentially influence the phenotype enough to influence its penetrance or expressivity. Environmental perturbations, such as maternal smoking, can also influence craniofacial phenotypes, for example, maternal smoking in the presence of the CA4 homozygous state increased the risk of clefting.19, 46 Thus, in the interest of future meta-analysis of these questions, we report these data below.
In summary, this report describes inheritance patterns and functional studies of an MSX1-coding mutation within its MH3 domain, identified in a three-generation Japanese family with mild hypodontia. The results of in vitro functional assays revealed that this amino-acid substitution partially reduces MSX1 protein transcriptional activity, but exhibits normal nuclear localization. Not surprisingly, neither the MSX1 CA repeat marker genotypes nor parental smoking status demonstrated discernible effects on tooth phenotype. Significantly, by targeting familial hypodontia, this report is the first to identify a moderately penetrant, likely low-frequency, population-specific, hypomorphic missense allele in the MSX1 gene co-segregating with this phenotype.
Subjects and methods
Patients
Seven families affected with hypodontia were investigated in this study. The diagnoses of all patients were based on X-rays. All individuals evaluated for either MSX1 or PAX9 mutations were of a sufficient age to establish their secondary tooth number phenotype. The control group comprised 50 Japanese samples from unrelated individuals who had no family history of hypodontia or facial malformation. A blood or hair sample was obtained with informed consent following Institutional Review Board approval in Aichi-Gakuin University, and the Institute for Developmental Research, Aichi Human Service Center.
Sequencing, mutation analysis of candidate genes
MSX1 and PAX9 genes were chosen for mutation screening. Primers were used according to previous reports,15, 47 and were used to amplify all exons of the candidate genes by PCR from genomic DNA. The MSX1 gene consists of two exons separated by a 1.6-kb intron that has a polymorphic CA dinucleotide repeat within its boundaries. The intronic CA repeat varies in length from 8 to 12 repeats that were originally defined as follows: CA1 (12 × CAs), CA2 (11 × CAs), CA3 (10 × CAs), CA4 (9 × CAs) and CA5 (8 × CAs). In the current version of the human genome sequence, UCSC Genome Browser on Human February 2009 (GRCh37/hg19) Assembly, 12 CA repeats are listed at genomic position chr4:4863624–4863648. Although http://www.hgvs.org/mutnomen/examplesDNA.htmlvar suggests usage of CA[4]+[2], for the sake of brevity, these CA repeat genotypes will be referenced in the text and figures either as CA4/2 or simply as 4/2. Primers and conditions for PCR amplification of the MSX1 CA dinucleotide repeat marker were as previously reported.17 PCR products were sequenced with the BigDye Terminator 2.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Sequencing reactions were size-separated on the ABI Prism 3100 DNA Analyzer (Applied Biosystems), and sequence data was collected with the ABI Data Collection software version 1.1 and subsequently analyzed with the ABI DNA Sequencing Analysis version 3.6.
Plasmid constructs and site-directed mutagenesis
DNA copies of exon 1 and exon 2 of human MSX1 were amplified from human genomic DNA with PCR. The FLAG epitope-coding nucleotide sequence was added at the 5′-terminus of exon 1. SalI restriction enzyme sites without amino-acid substitution were also generated at the 3′-terminus of exon 1 and at the 5′-terminus of exon 2 with/without the G>T transversion at nucleotide position 453 of the coding region, using PCR. These two fragments were subcloned into the pcDNA3 (Invitrogen, San Diego, CA, USA). The MyoD reporter plasmid (MyoD-Enhancer/Promoter-Luciferase)1, 48 was provided by Dr Cory Abate-Shen (Columbia University). Construct accuracy was confirmed by sequencing.
Cell culture and differentiation
A murine myoblast cell line, C2C12, was cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum and 5% (v/v) horse serum, at 37°C with 5% CO2. Cells were grown to 60% confluence and subcultured every 48 h in culture dishes. To induce myogenesis, the growth media was replaced with differentiation media (DMEM supplemented with 5% horse serum) after the cells reach 70% confluence, and the culture was continued for 3 days.
DNA transfection and determination of luciferase activity
The DNA transfection and luciferase assay were performed according to the previously reported method, with minor modifications.49 Briefly, cells were incubated in DMEM supplemented with 10% fetal bovine serum and 5% (v/v) horse serum containing plasmid DNA and Lipofectamine 2000. Each transfection point was carried out in triplicate in a gelatin-coated 24-well plate (Iwaki glass, Tokyo, Japan) and contained 1.0 μg of MyoD-luciferase reporter vector, 3–15 ng of MSX1 expression vectors and 3 ng of pRL-TK, an expression vector for the sea pansy (Renilla reniformis) luciferase used as an internal transfection control. The medium was subsequently replaced with DMEM supplemented with 5% horse serum, differentiation media after 22–26 h transfection and the cells were cultured for an additional 70–74 h. Firefly luciferase and Renilla luciferase activities were measured in a luminometer, using the Dual Luciferase Reporter assay system kit (Promega, Madison, WI, USA). Cells were lysed in 100 μl of passive lysis buffer in agitation for 30 min and 10 μl was used for measurement. Luciferase activity elicited by MyoD promoter constructs was normalized for variation in transfection efficiency using R. luciferase as an internal standard. The experiments were performed three times, and representative data are shown. Data are presented as mean and SD of the experiment. The comparison was carried out using Student's t-test. A P-value of <0.05 was considered to be statistically significant.
Western blotting
Western blotting was carried out in accordance with the protocol described previously, with minor modifications.50 In brief, transfectant cells in 35-mm dishes were rinsed twice with ice-cold phosphate-buffered saline (PBS). Lysis buffer (1% Triton X-100, 1-mM EDTA in Tris-buffered saline (TBS)); 20-mM Tris-HCl, pH 8.0 and 150-mM NaCl) with protease inhibitors was added to the cells. Cell lysates were maintained on ice for 30 min with occasional mixing and centrifuged (14 000 g for 15 min at 4°C); supernatants containing equal amounts of protein, as determined by the Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, USA), were subjected to PAGE (12% gel). Further, proteins were blotted from gels to Immobilon-P membranes (Millipore Corporation, Bedford, MA, USA). The blots were soaked for 1 h in blotting buffer (5% dried milk dissolved in TBS-T; TBS containing 0.1% Tween 20) to block nonspecific binding and washed with TBS. The membranes were incubated with anti-FLAG M2 monoclonal antibody (catalog no. F3165; Sigma, St Louis, MO, USA) at a dilution of 1:2000 in blotting buffer overnight at room temperature and then washed with TBS-T. The blots were incubated with horseradish peroxidase-conjugated secondary antibody in blotting buffer for 1 h at room temperature and the excess antibody was removed by washing in TBS-T. Finally, membranes were treated with enhanced chemiluminescence (ECL2) reagent (Amersham Life Science, Cleveland, OH, USA) and exposed to X-ray film.
Immunolocalization of the variant in mammalian cells
To investigate whether the R151S amino-acid substitution affects the subcellular localization of MSX1, transfection of C2C12 cells was carried out using Lipofectamine 2000 (Invitrogen, Grand Island, NY, USA). After 48-h transfection, cells were fixed with 4% paraformaldehyde/PBS and permeabilized with 1% Triton X-100/TBS before incubation with the anti-FLAG antibody (1:1000), for 60 min at 37°C. Further, cells were incubated for 60 min at room temperature with DAPI (1 μg/ml), Alexa-488 phalloidin (2 U/ml; Molecular Probes, Eugene, OR, USA) and Cy3-conjugated goat anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) (1:1000), respectively, in PBS. Further, after washing cells with PBS three times, immunostaining was visualized with an Olympus BH-2 microscope (Olympus, Tokyo, Japan).
Statistical analysis
Statistical analysis utilized the InStat 3 software (GraphPad Software, San Diego, CA, USA).
Results
Pedigree and diagnosis
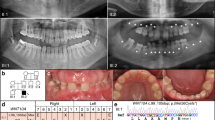
We examined the MSX1 and PAX9 exons, and flanking intronic nucleotide sequences of seven Japanese hypodontia families with at least two affected individuals (Figure 1 and Supplementary Figure 1). No orofacial cleft or any other craniofacial abnormalities were noted in any affected family members of the present study. In addition, all primary teeth were reported to be normal in size, shape and number. In one family, we identified a point mutation in the MSX1 gene (Figure 2a, right panel) of the proband. The severity of the phenotype in this family was quite mild, as the affected individuals had one or two missing permanent teeth, that is, hypodontia (Table 1). The proband (II-4) was an adult female, missing tooth ï (Palmer notation system). Her sister (II-2) was missing teeth ¥ and Jumlongras et al.3 Individuals III-1, lacking tooth ¢, and III-2, lacking teeth ¢ and °, were dizygotic twin daughters of II-2, who was married to an unaffected individual (II-1), not carrying a R151S mutation. Examination of the proband DNA from six other pedigrees revealed no additional DNA sequence variation in either MSX1 or PAX9. Two individuals were identified with a single peg-shaped tooth from among all these hypodontia families (Supplementary Figure 1).

Hypodontia family pedigree. (a) Pedigree analysis of hypodontia. Darkened symbols represent those individuals who had a hypodontia phenotype, whereas clear symbols indicate individuals with no tooth phenotype. Symbol in gray indicates that it was not possible to diagnose a tooth-loss phenotype. Squares indicate male and circles indicate females. An arrowhead indicates a cigarette smoker. DNA sequencing revealed that all family members with hypodontia and one with normal tooth numbers (II-3) carried a heterozygous R151S MSX1-coding substitution. The MSX1-R151S heterozygous genotype is followed by the MSX1 intronic CA repeat allele genotype, in which they were ascertained. (b) Hypodontia phenotype. Panoramic tomogram of a 27-year-old hypodontia female (patient II: 4) with only the lower left second premolar missing. The asterisk (*) indicates the mandibular region missing this secondary tooth.

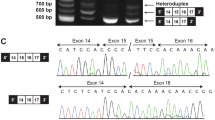
MSX1 R151S mutation identified in hypodontia family. (a) DNA sequence of part of exon 2 of MSX1 in unaffected (left panel; I-1) and affected (right panel; II-4) individuals. GT transversion at nucleotide position 453 in mutant sequence results in an amino-acid substitution in the MH3/extended homeodomain of MSX1. (b) Schematic representation of the human MSX1 protein, showing the location of the amino-acid substitution. (c) Multiple sequence alignment of MSX1 sequences sorted by species and homology. Only partial N-terminal sequences of the MH3/extended homeodomain are shown. Sequences were obtained from the SwissProt database and aligned. Amino-acid conservation among all species listed: arrow indicates conserved R151 with black shade; amino acid sequence differences from human MSX1 are marked with dark gray shade.
Mutation search of the family with hypodontia and MSX1 polymorphism
The proband from this family (II-4 in Figure 1) was shown to carry a heterozygous G>T transversion at nucleotide position 453 (as counted from the A of the MSX1 translational start codon; c.453G>T) in exon 2 of the affected individual in the family (Figure 2a, right panel, eg, I-1, shown in Figure 2a, left panel), but not found in 100 normal Japanese control chromosomes (not shown). This G>T transversion results in a replacement of arginine codon 151 by a serine codon (p.R151S) in the MH3 region of MSX11 (Figure 2b). The arginine in this position is a highly conserved amino acid within the broadly conserved MH3 domain (Figure 2c; but see the comprehensively curated alignment1). Further sequencing work revealed that four of five family members carrying the R151S variant were affected in this three-generation pedigree (Figure 1). No additional variants were uncovered. This nucleotide transversion was also absent in 500 control chromosomes from unrelated control individuals in multiple populations in a previous study,21 allowing an estimation of global allele frequency to be ≤0.01.
The 453 G>T transversion creating the R151S missense substitution is identical to the variant previously reported in a Japanese case proband with unilateral cleft lip and palate.21 The tooth phenotype of that individual was not ascertained at that time and further data are unfortunately no longer available.
R151S substitution partially reduces repression activity of MSX1 upon the MyoD-promoter in vitro
To test the consequences of the R151S missense variant upon MSX1 protein function, we choose to assess the output from an in vitro transcription assay, as justified in the discussion section. Thus, the transcriptional regulatory activity of the MSX1 variant was measured in C2C12 cells transfected with a reporter vector containing the MyoD promoter. Upon induction of differentiation, a considerable accumulation of the luciferase activity was measured in the control C2C12 cells without exogenous MSX1 expression (Figure 3a, left column in green). This activity is conspicuously reduced in the wild-type MSX1 transfectant in a dose-dependent manner. The blue columns of Figure 3a reveal increased repression (reading from left to right) with increased dose of wild-type human MSX1. The red columns of Figure 3a reveal loss of this repression activity when the variant MSX1-R151S was added to these assays. The loss of repression is significantly different from the amount induced by wild-type MSX1 at both 10 and 15 ng of added MSX1. In other words, significantly less repression was detected in R151S MSX1 variant-transfected cells as compared with those transfected with wild-type MSX1. To exclude the possibility that it was higher amounts of plasmid DNA that was adversely affecting luciferase activity, a control empty vector was assayed. No effect on MyoD activity was noted even when 20 ng/well of empty pcDNA3 control vector was added to the assay (without the MSX1 cDNA insert), thus allaying our concerns (data not shown). Several significant differences are highlighted with brackets and asterisks (*) in the figure. It is notable that with increasing dose of both wild-type and variant MSX1, increased repression was observed. This is obvious for those experiments that used the wild-type MSX1. It is subtler, but there was still significantly more repression demonstrated with increasing dose of the MSX1-R151S variant protein (with increases from 3- to 15-ng/well). This suggests that MSX1-R151S is a partially defective, hypomorphic mutation of the wild-type MSX1 protein, as it still retains some function in this assay.

MSX1 R151S functional assays. (a) C2C12 myoblasts were co-transfected with expression vectors for MSX1 and two reporter genes, the firefly luciferase reporter gene driven by the MyoD promoter and a sea pansy luciferase reporter gene driven by the TK promoter. After the treatments, firefly luciferase activity was measured and corrected with respect to sea pansy luciferase activity. Green column, without MSX1 expression; blue column, human wild-type MSX1 transfectant; and red, R151S variant of MSX1 transfectant. The experiments were performed three times and representative data are shown. Asterisks indicate that the P-value is <0.05, as determined by Student's t-test. (b) Western blot analysis was performed using extracts prepared from C2C12 cells transfected with a control vector or FLAG-Msx1 or FLAG-R151S-Msx1. The whole-cell extracts probed with the anti-FLAG antibody. Lane 1, human wild-type MSX1; lane 2, human R151S-MSX1. (c) Nuclear localization of MSX1 variant. The FLAG-tag attached to both the wild-type and R151S-MSX1 protein were detected by immunolocalization in the nucleus of transfected C2C12 cells at roughly equivalent levels. Mock-transfected C2C12 cells were used as negative control. Actin filament (phalloidin, green); MSX1 (FLAG, red); nuclei (DAPI, blue). White arrowheads indicate nuclei of transfectants. The R151S variant does not affect the nuclear localization potency of MSX1 in vitro. Scale bar=50 μm.
The stability of the variant mRNA and protein were, however, likely not affected by the nucleotide substitution, as we could not detect significant difference in protein expression level between the wild-type MSX1 and mutant R151S (Figure 3b).
Nuclear localization signal is not disrupted by the R151S mutation
It was considered possible that the reduced repression activity of the mutant R151S might be caused by the loss of nuclear localization. This was especially worrisome as the R151 is found within a region that apparently contains a bipartite nuclear localization signal, as previously suggested by virtue of its strong sequence alignment to and functional analysis of such a region within the Dlx3 protein.51 To test whether the R151S mutation affects nuclear localization of the protein, C2C12 cells were transfected with plasmids encoding FLAG-tagged wild-type or mutant MSX1. Using immunocytochemistry, we detected the signals for FLAG tag predominantly in the nucleus in both transfectants at 48 h after transfection (Figure 3c) and concluded that the variant does not appreciably affect nuclear localization.
MSX1 CA dinucleotide repeat genotyping and parental smoking status
The data above establish that the R151S is a moderately penetrant hypomorphic allele. Given that, this is the second report of this allele in the Japanese population, it may be that R151S is a low-frequency allele within that population with mild effects on the tooth-number phenotype. In this context, it becomes necessary to compare and contrast the mild influence of the R151S mutation with other mild complex disease alleles, such as the CA4 described above. These alleles could conceivably influence the variability of the phenotypes noted in our index family.
As cited above, although the CA4 allele is often associated with disease, the CA2 may be protective. Thus, we hypothesized, for example, that the CA2 allele might attenuate the effect of the amino-acid substitution in individual II-3, the only individual, carrying the R151S variation, who did not exhibit hypodontia (Figure 1a), and help to explain this difference in penetrance. Also noted above was the finding that these markers have demonstrated an association with susceptibility to environmental risk factors, such as parental smoking status. Thus, the CA repeat genotypes of all probands and all the available members of the R151S carrier family, as well as the parental smoking status, were ascertained and are listed in Figure 1, Supplementary Figure 1 and Table 2. However, there was no obvious correlation within this single family between the CA marker genotypes, smoking status and either the penetrance or expressivity of the phenotypes.
Discussion
Summary of findings
The coding mutations previously identified within patients exhibiting non-syndromic tooth agenesis were all inherited in a Mendelian manner. All those case studies reported oligodontia (loss of more than six secondary teeth, excluding the third molars). The present report, thus, represents the first description of an MSX1 missense mutation in a family exhibiting a consistent, yet slightly less penetrant, hypodontia phenotype. This tooth-loss pattern is consistent with previous reports of tooth agenesis among patients carrying MSX1 mutations, as well as with general tooth-loss patterns.3, 27, 52
Our in vitro functional studies of this missense polymorphism suggest that it is a hypomorphic mutation that is mildly deleterious, which still retains some repression activity at higher doses. This dose-dependent response was a very interesting result; especially as MSX1 effects are known to be dose sensitive, as previously cited.53 It is not known whether these experimental dose differences are developmentally relevant, but it does suggest how dose and function might interact to influence phenotype.
To justify our choice of the MyoD functional assay, several previous reports are revealing. For instance, it was previously reported that the MSX1 protein represses MyoD-responsive promoter activity, thereby maintaining mesenchymal cell proliferation.54, 55 A MyoD enhancer-promoter-luciferase reporter construct was subsequently used to demonstrate complete loss of repression function for the R196P MSX1 mutation identified in a large family displaying oligodontia.56 Although in vitro MyoD luciferase assay in C2C12 cells may not be the ideal assay to assess the function of MSX1 in tooth development, it did demonstrate that the MyoD reporter results were consistent with the biochemical data therein. Thus, this previous work established the relevance and appropriateness of using this assay again to evaluate the function of R151S mutation in dental and facial development. This current report also confirms previous bioinformatic analyses that suggested that R151S was a partially deleterious allele found at a highly conserved site, within the ancient, highly conserved MH3 domain of MSX1.1 Additional clinical and functional assays need to be developed before full understanding of all can be achieved.
Variable pleiotropy of MSX1 mutations
The 453 G>T transversion creating the R151S missense substitution is identical to the variant previously reported in a Japanese case proband with unilateral cleft lip and palate.21 The tooth phenotype of that individual was not ascertained at that time and further data are unfortunately no longer available. However, the second report of this particular nucleotide transversion in another Japanese individual suggests that R151S may be a population-specific allele. The lack of full phenotype ascertainment in the previous case report makes direct comparison problematic. Those individuals in the present family exhibiting hypodontia do not display any obvious craniofacial anomalies, such as oral facial clefting. Thus, we hypothesize that the clefting phenotype occurs only when additional loci besides the R151S allele at the MSX1 locus are disrupted. Yet, exactly how this genotype might contribute to the variable pleiotropy with such divergent phenotypes remains largely unanswered. It is of course still not possible at this time to generalize these findings toward other alleles at the MSX1 locus.
Low frequency, population-specific MSX1 alleles: common disease rare variants?
As the R151S variant has now been identified within a second Japanese individual, it raises the possibility that this will prove to be another low-frequency population-specific MSX1 allele. If this proves to be the case, then it would join other such candidate low-frequency, population-specific alleles, for example, E78V so far found only in Filipino populations,21 and P147Q identified in Vietnamese and Thai populations17, 22 and perhaps A34G in the Nigerian population in Africa (Butali et al., submitted). Although much more work remains before it is known whether these variants are pathogenic or merely neutral variants, given the high morbidity of the clefting phenotype, this remains an important question.
In fact, our preliminary estimate of the minor allele frequency of R151S in the global population is approximately ≤0.01, suggesting that it qualifies as a rare variant, as defined elsewhere.57 Increasingly, evidence points to a substantial role for rare, especially population-specific, allelic variants in common, complex diseases.58, 59, 60 It will be very interesting to pursue the incidence of the R151S mutation among other families with tooth agenesis and especially within the Japanese general population. This might be especially revealing as Japanese populations are known to have a higher incidence of tooth agenesis,26, 61 and Asian populations in general have a higher incidence of oral–facial clefting disorders.62 Therefore, although it is clear that the R151S is a mutation that causes hypodontia in at least one family, preliminary evidence suggests that it may also be influencing the risk of the relatively more common and complex tooth agenesis and oral–facial clefting birth disorders. Future research will continue to focus upon the R151S variant and the other bioinformatically prioritized set of MSX1 polymorphisms through deeper clinical and functional analyses. Additional clinical and functional assays need to be developed before full understanding of all can be achieved.
References
Finnerty JR, Mazza ME, Jezewski PA : Domain duplication, divergence, and loss events in vertebrate Msx paralogs reveal phylogenomically informed disease markers. BMC Evol Biol 2009; 9: 18.
Satokata I, Maas R : Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 1994; 6: 348–356.
Jumlongras D, Bei M, Stimson JM et al: A nonsense mutation in MSX1 causes Witkop syndrome. Am J Hum Genet 2001; 69: 67–74.
Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE : A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet 1996; 13: 417–421.
van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK : MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet 2000; 24: 342–343.
Lidral AC, Reising BC : The role of MSX1 in human tooth agenesis. J Dent Res 2002; 81: 274–278.
De Muynck S, Schollen E, Matthijs G, Verdonck A, Devriendt K, Carels C : A novel MSX1 mutation in hypodontia. Am J Med Genet A 2004; 128A: 401–403.
Kim JW, Simmer JP, Lin BP, Hu JC : Novel MSX1 frameshift causes autosomal-dominant oligodontia. J Dent Res 2006; 85: 267–271.
Mostowska A, Biedziak B, Trzeciak WH : A novel c.581C>T transition localized in a highly conserved homeobox sequence of MSX1: is it responsible for oligodontia? J Appl Genet 2006; 47: 159–164.
Chishti MS, Muhammad D, Haider M, Ahmad W : A novel missense mutation in MSX1 underlies autosomal recessive oligodontia with associated dental anomalies in Pakistani families. J Hum Genet 2006; 51: 872–878.
Xuan K, Jin F, Liu YL et al: Identification of a novel missense mutation of MSX1 gene in Chinese family with autosomal-dominant oligodontia. Arch Oral Biol 2008; 53: 773–779.
Bailleul-Forestier I, Berdal A, Vinckier F, de Ravel T, Fryns JP, Verloes A : The genetic basis of inherited anomalies of the teeth. Part 2: syndromes with significant dental involvement. Eur J Med Genet 2008; 51: 383–408.
Bailleul-Forestier I, Molla M, Verloes A, Berdal A : The genetic basis of inherited anomalies of the teeth. Part 1: clinical and molecular aspects of non-syndromic dental disorders. Eur J Med Genet 2008; 51: 273–291.
Polder BJ, Van't Hof MA, Van der Linden FP, Kuijpers-Jagtman AM : A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dent Oral Epidemiol 2004; 32: 217–226.
Lidral AC, Romitti PA, Basart AM et al: Association of MSX1 and TGFB3 with nonsyndromic clefting in humans. Am J Hum Genet 1998; 63: 557–568.
Fallin MD, Hetmanski JB, Park J et al: Family-based analysis of MSX1 haplotypes for association with oral clefts. Genet Epidemiol 2003; 25: 168–175.
Suzuki Y, Jezewski PA, Machida J et al: In a Vietnamese population, MSX1 variants contribute to cleft lip and palate. Genet Med 2004; 6: 117–125.
Modesto A, Moreno LM, Krahn K, King S, Lidral AC : MSX1 and orofacial clefting with and without tooth agenesis. J Dent Res 2006; 85: 542–546.
van den Boogaard MJ, de Costa D, Krapels IP et al: The MSX1 allele 4 homozygous child exposed to smoking at periconception is most sensitive in developing nonsyndromic orofacial clefts. Hum Genet 2008; 124: 525–534.
WHO: Global registry and data base on craniofacial anomailes. Report of a WHO Registry Meeting on Craniofacial Anomalies. Brazil: Baura, 2001; 12: 1–3.
Jezewski PA, Vieira AR, Nishimura C et al: Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet 2003; 40: 399–407.
Tongkobpetch S, Siriwan P, Shotelersuk V : MSX1 mutations contribute to nonsyndromic cleft lip in a Thai population. J Hum Genet 2006; 51: 671–676.
Wilkie AO : Pitfalls in the phylogenomic evaluation of human disease-causing mutations. J Biol 2009; 8: 26.
Schalk-van der Weide Y, Beemer FA, Faber JA, Bosman F : Symptomatology of patients with oligodontia. J Oral Rehabil 1994; 21: 247–261.
Arte S, Pirinen S : Hypodontia; in: Bloch-Zupan A (ed): Orphanet encyclopedia 2004.
Goya HA, Tanaka S, Maeda T, Akimoto Y : An orthopantomographic study of hypodontia in permanent teeth of Japanese pediatric patients. J Oral Sci 2008; 50: 143–150.
Vastardis H : The genetics of human tooth agenesis: new discoveries for understanding dental anomalies. Am J Orthod Dentofacial Orthop 2000; 117: 650–656.
Gorlin RJ, Cohen MM, Hennekam RCM : Syndromes of the Head and Neck. New York: Oxford University Press, 2001.
Das P, Hai M, Elcock C et al: Novel missense mutations and a 288-bp exonic insertion in PAX9 in families with autosomal dominant hypodontia. Am J Med Genet A 2003; 118A: 35–42.
Lammi L, Halonen K, Pirinen S, Thesleff I, Arte S, Nieminen P : A missense mutation in PAX9 in a family with distinct phenotype of oligodontia. Eur J Hum Genet 2003; 11: 866–871.
Zhao JL, Chen YX, Bao L, Xia QJ, Wu TJ, Zhou L : [Novel mutations of PAX9 gene in Chinese patients with oligodontia]. Zhonghua Kou Qiang Yi Xue Za Zhi 2005; 40: 266–270.
Kapadia H, Frazier-Bowers S, Ogawa T, D'Souza RN : Molecular characterization of a novel PAX9 missense mutation causing posterior tooth agenesis. Eur J Hum Genet 2006; 14: 403–409.
Ogawa T, Kapadia H, Feng JQ, Raghow R, Peters H, D'Souza RN : Functional consequences of interactions between Pax9 and Msx1 genes in normal and abnormal tooth development. J Biol Chem 2006; 281: 18363–18369.
Nakatomi M, Wang XP, Key D et al: Genetic interactions between Pax9 and Msx1 regulate lip development and several stages of tooth morphogenesis. Dev Biol 2010; 340: 438–449.
Peters H, Neubuser A, Kratochwil K, Balling R : Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 1998; 12: 2735–2747.
Kist R, Watson M, Wang X et al: Reduction of Pax9 gene dosage in an allelic series of mouse mutants causes hypodontia and oligodontia. Hum Mol Genet 2005; 14: 3605–3617.
Wang Y, Groppe JC, Wu J et al: Pathogenic mechanisms of tooth agenesis linked to paired domain mutations in human PAX9. Hum Mol Genet 2009; 18: 2863–2874.
Tríbulo C, Aybar MJ, Sánchez SS, Mayor R : A balance between the anti-apoptotic activity of Slug and the apoptotic activity of msx1 is required for the proper development of the neural crest. Dev Biol 2004; 275: 325–342.
Lallemand Y, Bensoussan V, Cloment CS, Robert B : Msx genes are important apoptosis effectors downstream of the Shh/Gli3 pathway in the limb. Dev Biol 2009; 331: 189–198.
Mitchell LE, Murray JC, O'Brien S, Christensen K : Evaluation of two putative susceptibility loci for oral clefts in the Danish population. Am J Epidemiol 2001; 153: 1007–1015.
Vieira AR, Orioli IM, Castilla EE, Cooper ME, Marazita ML, Murray JC : MSX1 and TGFB3 contribute to clefting in South America. J Dent Res 2003; 82: 289–292.
Slayton RL, Williams L, Murray JC, Wheeler JJ, Lidral AC, Nishimura CJ : Genetic association studies of cleft lip and/or palate with hypodontia outside the cleft region. Cleft Palate Craniofac J 2003; 40: 274–279.
Hewitt JE, Clark LN, Ivens A, Williamson R : Structure and sequence of the human homeobox gene HOX7. Genomics 1991; 11: 670–678.
Padanilam BJ, Stadler HS, Mills KA et al: Characterization of the human HOX 7 cDNA and identification of polymorphic markers. Hum Mol Genet 1992; 1: 407–410.
Letra A, Menezes R, Granjeiro JM, Vieira AR : Defining subphenotypes for oral clefts based on dental development. J Dent Res 2007; 86: 986–991.
Chevrier C, Bahuau M, Perret C et al: Genetic susceptibilities in the association between maternal exposure to tobacco smoke and the risk of nonsyndromic oral cleft. Am J Med Genet A 2008; 146A: 2396–2406.
Vieira AR, Meira R, Modesto A, Murray JC : MSX1, PAX9, and TGFA contribute to tooth agenesis in humans. J Dent Res 2004; 83: 723–727.
Lee H, Quinn JC, Prasanth KV et al: PIAS1 confers DNA-binding specificity on the Msx1 homeoprotein. Genes Dev 2006; 20: 784–794.
Nakayama A, Murakami H, Maeyama N et al: Role for RFX transcription factors in non-neuronal cell-specific inactivation of the microtubule-associated protein MAP1A promoter. J Biol Chem 2003; 278: 233–240.
Tokita Y, Keino H, Matsui F et al: Regulation of neuregulin expression in the injured rat brain and cultured astrocytes. J Neurosci 2001; 21: 1257–1264.
Bryan JT, Morasso MI : The Dlx3 protein harbors basic residues required for nuclear localization, transcriptional activity and binding to Msx1. J Cell Sci 2000; 113 (Part 22): 4013–4023.
Stockton DW, Das P, Goldenberg M, D'Souza RN, Patel PI : Mutation of PAX9 is associated with oligodontia. Nat Genet 2000; 24: 18–19.
Blin-Wakkach C, Lezot F, Ghoul-Mazgar S et al: Endogenous Msx1 antisense transcript: in vivo and in vitro evidences, structure, and potential involvement in skeleton development in mammals. Proc Natl Acad Sci USA 2001; 98: 7336–7341.
Bendall AJ, Ding J, Hu G, Shen MM, Abate-Shen C : Msx1 antagonizes the myogenic activity of Pax3 in migrating limb muscle precursors. Development 1999; 126: 4965–4976.
Peters H, Balling R : Teeth. Where and how to make them. Trends Genet 1999; 15: 59–65.
Hu G, Vastardis H, Bendall AJ et al: Haploinsufficiency of MSX1: a mechanism for selective tooth agenesis. Mol Cell Biol 1998; 18: 6044–6051.
McCarthy MI, Abecasis GR, Cardon LR et al: Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008; 9: 356–369.
Bodmer W, Bonilla C : Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 2008; 40: 695–701.
Maher B : Personal genomes: The case of the missing heritability. Nature 2008; 456: 18–21.
Manolio TA, Collins FS, Cox NJ et al: Finding the missing heritability of complex diseases. Nature 2009; 461: 747–753.
Niswander JD, Sujaku C : Congenital Anomalies of Teeth in Japanese Children. Am J Phys Anthropol 1963; 21: 569–574.
Weinberg SM, Neiswanger K, Martin RA et al: The Pittsburgh Oral-Facial Cleft study: expanding the cleft phenotype. Background and justification. Cleft Palate Craniofac J 2006; 43: 7–20.
Acknowledgements
This work was supported in part by grants from the Institute for Developmental Research, Aichi-Human Service Center (Y Tokita), and from the Aichi-Gakuin University, School of Dentistry (K Shimozato). We thank Dr Cory Abate-Shen (Columbia University, USA) for providing us with the MyoD reporter plasmid. The work presented here was carried out in collaboration among all authors. J Machida, K Shimozato and Y Tokita defined the research theme. Y Higashi, A Nakayama, T Ono, PA Jezewski and Y Tokita designed methods and experiments. M Kamamoto, S Yamaguchi, M Kimura and Y Tokita carried out the laboratory experiments, and analyzed the data. PA Jezewski, and Y Tokita discussed and interpreted the final data set and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Kamamoto, M., Machida, J., Yamaguchi, S. et al. Clinical and functional data implicate the Arg(151)Ser variant of MSX1 in familial hypodontia. Eur J Hum Genet 19, 844–850 (2011). https://doi.org/10.1038/ejhg.2011.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.47
Keywords
This article is cited by
-
Novel frameshift variant of WNT10A in a Japanese patient with hypodontia
Human Genome Variation (2024)
-
Novel WNT10A variant in a Japanese case of nonsyndromic oligodontia
Human Genome Variation (2023)
-
A novel LRP6 variant in a Japanese family with oligodontia
Human Genome Variation (2021)
-
Novel MSX1 frameshift mutation in a Japanese family with nonsyndromic oligodontia
Human Genome Variation (2021)
-
A novel mutation of MSX1 in oligodontia inhibits odontogenesis of dental pulp stem cells via the ERK pathway
Stem Cell Research & Therapy (2018)

{kind=link}