
Abstract
Insertion sequencing (INSeq) is a method for determining the insertion site and relative abundance of large numbers of transposon mutants in a mixed population of isogenic mutants of a sequenced microbial species. INSeq is based on a modified mariner transposon containing MmeI sites at its ends, allowing cleavage at chromosomal sites 16–17 bp from the inserted transposon. Genomic regions adjacent to the transposons are amplified by linear PCR with a biotinylated primer. Products are bound to magnetic beads, digested with MmeI and barcoded with sample-specific linkers appended to each restriction fragment. After limited PCR amplification, fragments are sequenced using a high-throughput instrument. The sequence of each read can be used to map the location of a transposon in the genome. Read count measures the relative abundance of that mutant in the population. Solid-phase library preparation makes this protocol rapid (18 h), easy to scale up, amenable to automation and useful for a variety of samples. A protocol for characterizing libraries of transposon mutant strains clonally arrayed in a multiwell format is provided.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others
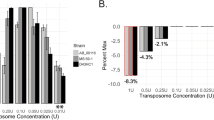
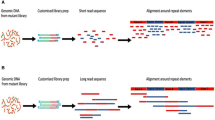
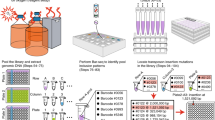
References
Mazurkiewicz, P., Tang, C.M., Boone, C. & Holden, D.W. Signature-tagged mutagenesis: barcoding mutants for genome-wide screens. Nat. Rev. Genet. 7, 929–939 (2006).
Goodman, A.L. et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289 (2009).
Erlich, Y. et al. DNA Sudoku—harnessing high-throughput sequencing for multiplexed specimen analysis. Genome Res. 19, 1243–1253 (2009).
Prabhu, S. & Pe'er, I. Overlapping pools for high-throughput targeted resequencing. Genome Res. 19, 1254–1261 (2009).
Xin, X. et al. Shifted Transversal Design smart-pooling for high coverage interactome mapping. Genome Res. 19, 1262–1269 (2009).
Gawronski, J.D., Wong, S.M., Giannoukos, G., Ward, D.V. & Akerley, B.J. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc. Natl. Acad. Sci. USA 106, 16422–16427 (2009).
Langridge, G.C. et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 19, 2308–2316 (2009).
van Opijnen, T., Bodi, K.L. & Camilli, A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–772 (2009).
Gallagher, L.A., Shendure, J. & Manoil, C. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. MBio 2, 00315-10 (2011).
van Opijnen, T. & Camilli, A. Genome-wide fitness and genetic interactions determined by Tn-seq, a high-throughput massively parallel sequencing method for microorganisms. Curr. Protoc. Microbiol. 19, 1E.3.1–1E.3.16 (2010).
Morgan, R.D., Bhatia, T.K., Lovasco, L. & Davis, T.B. MmeI: a minimal type II restriction-modification system that only modifies one DNA strand for host protection. Nucleic Acids Res. 36, 6558–6570 (2008).
Brody, J.R. & Kern, S.E. Sodium boric acid: a Tris-free, cooler conductive medium for DNA electrophoresis. Biotechniques 36, 214–216 (2004).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Westover, B.P., Buhler, J.D., Sonnenburg, J.L. & Gordon, J.I. Operon prediction without a training set. Bioinformatics 21, 880–888 (2005).
Acknowledgements
This work was supported by grants from the National Institutes of Health (DK30292, DK70977, and DK064540 to J.I.G.; F32AI078628 and K01DK089121 to A.L.G.) and by the Crohn's and Colitis Foundation of America. We thank L. Kyro and J. Eberle for their assistance in making the movies that accompany this article.
Author information
Authors and Affiliations
Contributions
A.L.G., J.I.G. and M.W. designed the experiments and software. A.L.G., M.W. and J.I.G. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Table 1
Primer sequences. (DOCX 137 kb)
Supplementary Dataset 1
INSeq_analysis.zip: INSeq data analysis pipeline and README.txt file. (ZIP 14615 kb)
Supplementary Movie 1
Pipetting technique for bead-based separation. This video demonstrates the process of removing the supernatant from beads with a multichannel pipettor as described in Step 34. (MOV 25545 kb)
Supplementary Movie 2
Mapping an arrayed INSeq library by combinatorial pooling. This video demonstrates the process of pooling strains using an EpMotion liquid handling robot as described in Box 1, Step 5. (MOV 25601 kb)
Rights and permissions
About this article
Cite this article
Goodman, A., Wu, M. & Gordon, J. Identifying microbial fitness determinants by insertion sequencing using genome-wide transposon mutant libraries. Nat Protoc 6, 1969–1980 (2011). https://doi.org/10.1038/nprot.2011.417
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2011.417
This article is cited by
-
Genome-wide identification of bacterial colonization and fitness determinants on the floating macrophyte, duckweed
Communications Biology (2022)
-
SorTn-seq: a high-throughput functional genomics approach to discovering regulators of bacterial gene expression
Nature Protocols (2021)
-
Transposon sequencing analysis of Bradyrhizobium diazoefficiens 110spc4
Scientific Reports (2021)
-
Interplay between Yersinia pestis and its flea vector in lipoate metabolism
The ISME Journal (2021)
-
Pneumococcal capsule blocks protection by immunization with conserved surface proteins
npj Vaccines (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
