
Abstract
The evolutionarily conserved target of rapamycin complex 1 (TORC1) is a master regulator of cell growth and metabolism. In mammals, growth factors and cellular energy stimulate mTORC1 activity through inhibition of the TSC complex (TSC1-TSC2-TBC1D7), a negative regulator of mTORC1. Amino acids signal to mTORC1 independently of the TSC complex. Here, we review recently identified regulators that link amino acid sufficiency to mTORC1 activity and how mutations affecting these regulators cause human disease.
Similar content being viewed by others

Introduction
Nutrients, growth factors and cellular energy trigger synthesis of essential building blocks, such as proteins, lipids, and nucleotides, and thereby stimulate growth. Target of rapamycin complex 1 (TORC1) integrates the above three inputs to activate the metabolic pathways that ultimately drive cell and organismal growth (reviewed in1,2,3,4,5,6,7,8). Mammalian TORC1 (mTORC1) activity is often deregulated in pathophysiological conditions such as diabetes and cancer (reviewed in9).
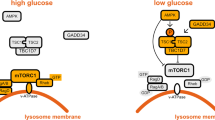
mTORC1 consists of mTOR, regulatory associated protein of mTOR (RAPTOR), and mammalian lethal with SEC13 protein 8 (mLST8)10,11,12, and is acutely sensitive to the macrolide rapamycin. Growth factors and cellular energy control mTORC1 via a heterotrimeric TSC complex consisting of tuberous sclerosis complex 1 (TSC1), TSC2, and TRE2-BUB2-CDC16 domain family member 7 (TBC1D7)13,14,15,16,17,18 (Figure 1). Binding of growth factors such as insulin to receptor tyrosine kinases (RTKs) activates phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) to generate phosphoinositide 3, 4, 5-phosphate (PIP3). PIP3 recruits phosphoinositide-dependent kinase 1 (PDK1) and AKT to the plasma membrane via plekstrin homology (PH) domains in these kinases (reviewed in19). PDK1 phosphorylates the activation loop (Thr308) in AKT and thereby activates AKT20,21. AKT phosphorylates TSC2 to inhibit the TSC complex by inducing its release from the lysosome22,23,24. The TSC complex is a GTPase-activating protein (GAP) toward the lysosomal, small GTPase RAS homologue enriched in brain (RHEB). GTP-loaded RHEB activates mTORC1 via direct interaction with the mTOR catalytic domain25, but the precise mechanism of mTORC1 activation by RHEB awaits structural analysis of mTORC1 in complex with RHEB. A high AMP/ATP ratio (low cellular energy) activates AMP-dependent kinase (AMPK) to phosphorylate TSC2, at sites distinct from AKT target sites, and to stimulate GAP activity of the TSC complex26. In contrast to growth factors and energy, nutrients (in particular amino acids) activate mTORC1 via a mechanism independent of the TSC complex. Amino acids also stimulate TORC1 activity in the yeast Saccharomyces cerevisiae that lacks a TSC complex. Unlike the insulin-AKT-TSC pathway, the amino acid-sensing branch of TORC1 is evolutionarily conserved from yeast to human. As described below, this branch consists of signaling through RAG GTPases.

Regulation of mTORC1. mTORC1 is activated by growth factors (such as insulin), cellular energy (ATP), and nutrients (amino acids). Growth factors activate the PI3K-PDK1-AKT pathway to inhibit the TSC complex, a GAP for RHEB. Upon inhibition of the TSC complex, GTP-bound RHEB binds and activates mTORC1 on the lysosome. Low cellular energy (high AMP/ATP ratio) stimulates AMPK to phosphorylate and activate the TSC complex, thus inhibiting RHEB and mTORC1. Amino acids promote activation of the RAG proteins to recruit mTORC1 to the lysosomal surface where it encounters RHEB. The GTPase RAGA or RAGB (RAGA/B) forms a heterodimer with the GTPase RAGC or RAGD (RAGC/D). Amino acids activate the RAG heterodimer by modulating its guanine nucleotide binding status. The active RAG heterodimer contains GTP-bound RAGA/B and GDP-bound RAGC/D. The heteropentameric RAGULATOR complex anchors the RAGs to the lysosomal surface, and is a GEF for RAGA/B. Upstream of RAGULATOR/RAGs, glutaminolysis promotes GTP loading of RAGB. The guanine nucleotide status of RAGA/B is also regulated by the GATOR1 complex (GAP) and its negative regulator GATOR2. SESN2 is a negative regulator of both GATOR2 and RAGA/B, in the latter case as a GDI. SESN2 is reported to be a cytoplasmic leucine sensor. Leucine binds to SESN2 and inhibits SESN2-GATOR2 interaction. The FLCN-FNIP complex is a GAP for RAGC/D. SLC38A9 is an arginine transporter in the lysosomal membrane and interacts with the RAGs and RAGULATOR to activate mTORC1. Glutamine also promotes lysosomal translocation and activation of mTORC1 through ARF1 in a RAG-independent manner. V-ATPase is required for lysosomal recruitment and activation of mTORC1 in RAG-dependent and -independent manners. Amino acids also activate mTORC1 via recruitment to the Golgi, involving RAB1A and Golgi-resident RHEB. Phosphorylation represented in yellow and red indicates an activation and inhibitory signal, respectively. Arrows and bars represent activation and inhibition, respectively, of the downstream protein.
mTORC1 phosphorylates ribosomal protein S6 kinase (S6K) and eukaryotic translation initiation factor 4E-binding protein (4E-BP) to promote anabolic processes such as protein, lipid, and nucleotide synthesis, and unc-51 like autophagy activating kinase 1 (ULK1) to inhibit autophagy, leading to cell growth. Furthermore, mTORC1 and S6K phosphorylate growth factor receptor-bound protein 10 (GRB10) and insulin receptor substrate 1 (IRS1), respectively27,28,29. Phosphorylation of GRB10 and IRS1 inhibits PI3K activation, thus forming a negative feedback loop that has an important role in the regulation of growth factor signaling27,28,29.
Biochemical and genetic studies have identified upstream regulators of mTORC1 in the amino acid-sensing pathway (Figure 1). However, the precise mechanism by which amino acid availability is transduced to mTORC1 and how different amino acids promote mTORC1 activity are still elusive. In this review, we discuss the amino acid sensing pathway in the regulation of mTORC1.
RAG-dependent amino acid signaling to mTORC1
RAG
How amino acids signal to mTORC1 is a long-standing question. Hara et al.30 demonstrated that phosphorylation of mTORC1 targets S6K and 4E-BP decreases after withdrawal of amino acids, especially leucine, and that insulin fails to stimulate S6K and 4E-BP phosphorylation in the absence of amino acids. These findings suggest that amino acids and insulin act on mTORC1 via distinct mechanisms, both of which are necessary to activate mTORC1. In contrast to the growth factor-sensing insulin-PI3K-AKT pathway, components of the amino acid-sensing branch of mTORC1 were revealed only recently. A first hint came from a genome-wide screen in yeast31. Dubouloz et al.31 reported that the RAG super family member Gtr2, which forms the EGO complex with Ego1, Ego2, and Ego332, is required for exit from rapamycin-induced growth arrest. In addition, cells deficient for TORC1 activity (tor1Δ) are synthetically sick upon deletion of GTR2, indicating a genetic interaction between TOR1 and GTR2. However, this study did not investigate the role of Gtr2 or the EGO complex in the context of a nutrient-sensing pathway. In 2008, two independent groups identified RAG small GTPases as essential components in an amino acid-sensing pathway upstream of TORC1 in flies and mammalian cells33,34 (Figure 1). Sancak et al.34 biochemically purified RAPTOR and analyzed associated proteins by mass spectrometry (MS). Kim et al.33 screened small GTPases in insect cells to identify components of an amino acid-sensing pathway upstream of TORC1. In mammals, there are four RAGs35,36,37. The highly homologous RAGA or RAGB forms a heterodimer with RAGC or RAGD. Amino acids convert the RAG heterodimer to its active conformation in which RAGA or RAGB is loaded with GTP (RAGA/B-GTP) and RAGC or RAGD is loaded with GDP (RAGC/D-GDP). Sancak et al.34 further demonstrated that the active RAG heterodimer binds RAPTOR without affecting mTORC1 kinase activity. Instead, the active RAG heterodimer recruits mTORC1 to the surface of the lysosome where RHEB binds and activates mTORC1. In the absence of amino acids, mTORC1 is dispersed in the cytoplasm. It is unclear whether dispersed mTORC1 binds to vesicles or exists as a free complex. Importantly, Sancak et al.34 answered the long-standing question of why neither amino acids nor growth factors alone are sufficient to activate mTORC130,38. Shortly thereafter, Binda et al.39 showed that the RAG family proteins in yeast, Gtr1 and Gtr2, are also activators of TORC1 in response to leucine (Figure 2). Gtr1 is an ortholog of RAGA and RAGB whereas Gtr2 is an ortholog of RAGC and RAGD36,40. Like RAG proteins in flies and mammals, Gtr1 and Grt2 form a heterodimer complex in which the active conformation is Gtr1-GTP in complex with Gtr2-GDP. Leucine promotes the loading of relevant guanine nucleotides on Gtrs and thereby facilitates a direct physical interaction between TORC1 and the Gtr heterodimer. More recently, Valbuena et al.41 demonstrated that Gtr1 and Gtr2 are upstream regulators of TORC1 in fission yeast. In contrast to mammals, TORC1 in yeast is localized to the vacuole (yeast equivalent of the lysosome) regardless of leucine availability or guanine nucleotide status of the Gtr proteins. It is unclear how the Gtr heterodimer activates TORC1 or how TORC1 is localized to the vacuole in yeast.

Regulation of TORC1 in yeast. Yeast TORC1 localizes to the vacuolar surface. The yeast vacuole is equivalent to the mammalian lysosome. Gtr1 and Gtr2 are orthologs of RAGA/B and RAGC/D, respectively. Like RAGs, Gtr1 and Gtr2 form a heterodimer, of which the active conformation is Gtr1-GTP and Gtr2-GDP. As part of the EGO complex, Gtr1 and Gtr2 are anchored to the vacuole via Ego1, Ego2, and Ego3. Leucine promotes GTP loading of Gtr1, which depends on the GEF Vam6 or the moonlighting leucyl tRNA synthetase Cdc60. The trimeric SEACIT, the yeast GATOR1 ortholog, is a GAP for Gtr1 and is inhibited by the pentameric SEACAT, the yeast GATOR2 ortholog. The Lst4-Lst7 complex is a GAP for Gtr2 downstream of glutamine. Glutamine also mediates sustained TORC1 activity independently of the Gtr proteins. Arrows and bars represent activation and inhibition, respectively, of the downstream protein.
Demetriades et al.42 demonstrated that amino acid starvation in the presence of growth factors leads to “inactive” RAGs that in turn bind and recruit the TSC complex to the lysosome to inhibit RHEB and thereby TORC1. However, Menon et al.24 demonstrated that lysosomal translocation of the TSC complex is sensitive to growth factors, and not to amino acids. The reason for this discrepancy is unknown.
RAGULATOR, a GEF for RAGA/B
RAG proteins localize to the lysosomal surface in an amino acid-independent manner, although they do not possess a transmembrane domain or a motif predicted to be lipid-modified. This suggests that another lysosomal protein is involved in the lysosomal localization of the RAGs and mTORC1. Following a biochemical approach similar to the one that identified the RAGs, two studies identified a protein complex consisting of p18 (later renamed LAMTOR1), p14 (LAMTOR2), MP1 (LAMTOR3), C7orf59 (LAMTOR4), and HBXIP (LAMTOR5), thereafter known as the RAGULATOR complex38,43. Like the RAGs, the RAGULATOR complex localizes to the lysosome in an amino acid-independent manner. Knockdown of any of the components of the RAGULATOR complex abrogates lysosomal localization of the RAGs and mTORC1 as well as mTORC1 activity. Importantly, LAMTOR1 is both myristoylated and palmitoylated, and these lipid modifications are required for LAMTOR2 and LAMTOR3 localization to the lysosome44. Thus, the RAGULATOR complex serves as a docking site for RAGs and mTORC1 on the lysosome.
RAGs are small GTPases whose ability to regulate TORC1 depends on their GTP- or GDP-loading status. Importantly, amino acids control the guanine nucleotide status of RAGs. In general, exchange of GDP for GTP in a small GTPase is promoted by a guanine nucleotide exchange factor (GEF). Conversely, small GTPases have weak intrinsic GTP hydrolysis activity, and thus conversion of GTP to GDP requires a GTPase-activating protein (GAP). Bar-Peled et al.43 demonstrated that the pentameric RAGULATOR complex, but not any single subunit, possesses GEF activity toward RAGA and RAGB, and possibly toward RAGC. It is unknown which subunit(s) in RAGULATOR has GEF activity. As discussed in the study of Bar-Peled et al.43, the entire pentameric complex might be required for GEF activity. Structural analysis of RAGULATOR may identify the GEF site in the pentameric complex. A structural analysis might also explain the stronger GEF activity toward RAGA/B versus RAGC/D.
Although there is no RAGULATOR ortholog in yeast based on primary sequence, a recent study revealed that the yeast Ego1-Ego2-Ego3 ternary complex (EGO complex) is a structural ortholog of the RAGULATOR complex32 (Figures 1 and 2). Ego1 contains potential myristoylation and palmitoylation sites at its N-terminus, like LAMTOR1, and deletion of EGO1 abrogates vacuolar localization of Ego2 and Ego332,45,46. An N-terminal region of Ego3 serves as a docking site for Gtr1-Gtr2 on the vacuole. However, it is unclear whether the EGO complex has GEF activity toward Gtr1. Binda et al.39 identified Vam6 in a genome-wide screen as a potential GEF for Gtr1 (Figure 2). Vam6 physically interacts with Gtr1, and stimulates GDP release from Gtr1 in vitro, indicating that Vam6 is indeed a GEF for Gtr1. VPS39 is the mammalian ortholog of yeast Vam6, but VPS39 does not interact with RAGA and has no effect on guanine nucleotide release from RAGB, suggesting that VPS39 is not the GEF for RAGA and RAGB in mammals43.
Vacuolar H+-ATPase (V-ATPase)
The identification of the lysosome as a platform for mTORC1 suggested that there might be other lysosomal components involved in the amino acid-sensing branch of mTORC1. An RNA interference screen targeting lysosomal proteins in Drosophila S2 cells revealed that knockdown of genes encoding the vacuolar H+-ATPase (V-ATPase) subunits inhibits amino acid-stimulated S6K phosphorylation47 (Figure 1). V-ATPase is an ATP-driven proton pump that maintains cytoplasmic pH by acidifying the lysosomal lumen. It is a multi-subunit complex consisting of an ATP-hydrolytic domain (V1) and an integral membrane proton-pumping domain (V0) (reviewed in48). Pharmacological inhibition of V-ATPase or knockdown of genes encoding V-ATPase components also prevents amino acid-induced mTORC1 activation47. Furthermore, Gtr1 interacts with V-ATPase in yeast, and deletion of a V-ATPase component (vma2Δ) inhibits TORC1 activity49. These data suggest that the V-ATPase is an evolutionarily conserved upstream regulator of TORC1. Although it is not clear whether the V-ATPase in yeast is required for TORC1 activation in response to amino acids, this is likely the case since the growth defect of vma2Δ cells is partially reversed by overexpression of Gtr1-GTP49.
How does V-ATPase promote mTORC1 activity? Biochemical analysis demonstrated that the RAGULATOR complex physically interacts with V-ATPase via LAMTOR1-V0 domain and LAMTOR2-V1 domain associations. The interaction between LAMTOR2 and the V1 domain, but not the LAMTOR1-V0 interaction, is sensitive to amino acids, i.e., amino acids weaken the interaction47. The physiological significance of this regulated interaction is unknown. In cells expressing a constitutively active form of RAGA or RAGB, mTORC1 is constitutively localized to the lysosome and is active even in the presence of V-ATPase inhibitors47. Thus, V-ATPase appears to act upstream of the RAG and RAGULATOR complexes and is required for lysosomal translocation of mTORC1 in response to amino acids via an unknown mechanism.
GATOR1, a GAP for RAGA/B
Two studies independently identified the GAPs for yeast Gtr1 and mammalian RAGA/B50,51. In yeast, Iml1 forms a complex with Npr2 and Npr3 (a complex termed SEACIT, Seh1-associated complex subcomplex inhibiting TORC1 signaling)52,53,54. Iml1 in the SEACIT complex acts as a GAP toward Gtr1. Mutation of the GAP domain in Iml1 abolishes the GAP activity and ability to inhibit TORC151 (Figure 2). The mammalian orthologs of SEACIT components, DEP domain containing 5 (DEPDC5), nitrogen permease regulator 2-like protein (NPRL2), and NPRL3, form a complex named GATOR1 that has GAP activity toward RAGA/B50 (Figure 1). mTORC1 is insensitive to amino acid withdrawal in GATOR1-depleted cells50. The GAP domain in yeast Iml1 is conserved in mammalian DEPDC551. Expression of human DEPDC5 in iml1Δ cells suppressed TORC1 activity, suggesting that DEPDC5 in GATOR1 is the GAP catalytic subunit toward RAGA/B-GTP51.
The yeast Seh1-associated complex subcomplex activating TORC1 (SEACAT), consisting of Sec13, Seh1, Sea2, Sea3, and Sea4, and the mammalian GATOR2, a complex consisting of SEC13, SEH1L, WDR24, WDR59, and MIOS, are interactors and negative regulators of SEACIT and GATOR1, respectively54. The reader is referred to Dokudovskaya and Rout55 for more details on SEA and GATOR complexes.
SESTRIN
SESTRIN (SESN)1/2/3 are stress-inducible proteins that maintain metabolic homeostasis and suppress obesity- and age-mediated pathologies (reviewed in56). It was previously reported that SESNs inhibit mTORC1 through activation of AMPK and the TSC complex, causing inhibition of cell growth57,58. More recently, four independent studies demonstrated that SESNs inhibit mTORC1 independently of the AMPK-TSC complex axis59,60,61,62 (Figure 1). All four studies showed that SESNs, especially SESN2, inhibit amino acid-induced lysosomal localization and activation of mTORC1. However, two distinct mechanisms are described. Chantranupong et al.60, Parmigiani et al.61, and Kim et al.62 reported that SESNs bind and inhibit GATOR2. These studies also found that the interaction between SESNs and GATOR2 is strengthened upon amino acid starvation. Two of the studies showed that SESN overexpression does not affect the GATOR2-GATOR1 interaction or GAP activity of GATOR1 toward RAGA/B60,61. However, Kim et al.62 demonstrated that SESN2 overexpression indeed releases GATOR1 from GATOR2-mediated inhibition, thus allowing GATOR1 to stimulate hydrolysis of GTP in RAGB. Kim et al. speculated that this discrepancy could be due to the extent of SESN overexpression relative to expression of the GATOR complexes. Intriguingly, Kim et al. demonstrated that SESN2 overexpression or ER stress, a known inducer of SESN expression, induces dissociation of endogenous GATOR2 from endongenous GATOR1 in mouse liver. This finding suggests that SESN2 acts to release GATOR1 from GATOR2 in a physiologically relevant manner. Contrary to a mechanism in which SESNs act on GATOR2, Peng et al.59 suggested that SESN is a guanine nucleotide dissociation inhibitor (GDI) for RAGA and RAGB. Peng et al. further showed that SESNs interact with the RAGs and prevent GDP dissociation in RAGA and B, but not in RAGC or RAGD. Introduction of point mutations in the conserved GDI motif of SESN2 abolished its GDI activity toward RAGB and the ability of SESN2 to inhibit mTORC1 lysosomal localization and activity, supporting their hypothesis that SESNs act upstream of mTORC1 as GDIs for RAGA or RAGB. Peng et al.59 also showed that overexpressed SESN2 partially co-localizes with RAGs, presumably on the lysosome, while Parmigiani et al.61 and Kim et al.62 reported that overexpressed SESN2 does not localize to the lysosome. The reason for this discrepancy is not known. Investigation of endogenous SESN localization may clarify whether SESNs are localized to the lysosome to act as GDIs. The nature of the selectivity of GDI function for RAGA/B is unclear, but the study of Peng et al. speculated that negatively charged aspartic acid in the switch2 motifs of RAGA and RAGB interacts with positively charged amino acid sequences in the GDI motif of SESNs. Structural information on SESNs and the RAG heterodimer will provide further insight regarding the selectivity of SESN GDI activity. How do amino acids regulate the GDI activity of SESNs? Peng et al. speculated that amino acid stimulation promotes dissociation of SESNs from RAGA and RAGB through an unknown GDI displacement factor (GDF). As the authors further speculated, RAGULATOR may function as the GDF since GEFs can function as a GDF for their target GTPases. Alternatively, given the fact that GATOR2 interacts with SESNs in an amino acid-sensitive manner, GATOR2 might inhibit GDI activity of SESNs, a function distinct from negative regulation of GATOR160,61,62. At this point, both models, in which SESNs act as a negative regulator of GATOR2 and as a GDI for RAGA/B, are possible.
It is unknown whether a GDI for Gtr1 exists in yeast. Based on primary amino acid sequence, SESN orthologs are not found in yeast despite the extensive conservation of the TORC1 amino acid-sensing pathway. It is possible that SESNs are metazoan-specific regulators of mTORC1 in response to amino acids. Alternatively, like RAGULATOR and the EGO complex, a yet to be identified functional ortholog(s) may exist in yeast.
RNF152 and SKP2, E3 ubiquitin ligases for RAGA
It was recently reported that RAGA is also regulated by lysine 63-linked ubiquitination. Two independent studies demonstrated that RAGA ubiquitination promotes its binding to GATOR1, leading to inactivation of RAGA63,64. Deng et al.64 identified RNF152 as the E3 ubiquitin ligase that mediates ubiquitination of RAGA at lysine 142, 220, 230, and 244, while Jin et al.63 found that another E3 ubiquitin ligase, SKP2, mediates ubiquitination of RAGA at lysine 15. Each study showed that the described ubiquitination increases interaction between RAGA and GATOR1, and overexpression of a RAGA ubiquitination-deficient mutant (K142R, K220R, K230R, K244R, or K15R) reduces RAGA-GATOR1 interaction, and thus increases mTORC1 activity. How are RNF152 and SKP2 regulated in response to amino acids? Deng et al.64 showed that amino acid starvation increases RNF152 interaction with RAGA and RAGA ubiquitination. However, RNF152 preferentially binds to the already inactive RAGA-GDP, suggesting that RNF152-mediated ubiquitination is not the primary mechanism of RAGA inactivation by GATOR1. Jin et al.63 surprisingly found that amino acids promote SKP2-mediated RAGA interaction with GATOR1. This observation is inconsistent with a previous study showing that amino acid starvation increases the interaction between GATOR1 and RAGA50. SKP2 recruitment to RAGA upon amino acid stimulation is dependent on mTORC1 activity since rapamycin prevents SKP2 recruitment as well as interaction between RAGA and GATOR163. Jin et al. proposed that SKP2-mediated RAGA ubiquitination is a negative feedback mechanism to prevent mTORC1 hyperactivation upon amino acid stimulation. Both studies showed that MEFs from RNF152- or SKP2-knockout mice have increased mTORC1 activity, presumably due to increased RAGA-GTP levels63,64. Curiously, both RNF152 and SKP2 knockout mice are viable64,65 in contrast to neonatal lethality conferred by RAGA-GTP knockin66 or SESN knockout59 (see below). This may suggest that RNF152 and SKP2 have a small role in mTORC1 physiology, or their function may be relevant only in specific physiological contexts.
Folliculin (FLCN), a GAP for RAGC and RAGD
Compared to RAGA/B, regulation of the guanine nucleotide status in RAGC/D is less characterized. A GEF for RAGC/D or Gtr2 has yet to be identified. FLCN has been linked to the TOR pathway in different organisms including yeast67,68, fly69, mouse70,71,72, and human70,72,73. Two recent studies showed that FLCN in complex with FLCN-interacting protein (FNIP) promotes mTORC1 lysosomal translocation and activity in response to amino acids74,75 (Figure 1). FLCN localizes to the lysosome upon amino acid starvation and only when RAGA/B is inactive. Upon amino acid stimulation, RAGA or RAGB is loaded with GTP, liberating FLCN from the lysosome. Tsun et al.75 further demonstrated that FLCN is a GAP for RAGC and RAGD to promote the active conformation of RAGC and RAGD. Since FLCN is already localized to the lysosome in the absence of amino acids, it requires amino acids only to stimulate its GAP activity. Interestingly, FLCN is well conserved from yeast to human, and deletion of the yeast FLCN ortholog Lst7 is synthetically lethal with mutation of the sec13 gene76 encoding a component of SEACAT/GATOR254. An explanation for the synthetic lethality could be that double mutation of SEC13 and LST7 completely abolishes amino acid input into TORC1. Indeed, a recent study showed that Lst7 forms a complex with Lst4, a yeast ortholog of FNIP, and is a GAP for Gtr277. Like in mammals, the Lst4-Lst7 complex in yeast localizes to the vacuole upon amino acid starvation. Amino acids, especially glutamine, promote binding of the Lst4-Lst7 complex to Gtr2, hydrolysis of GTP in Gtr2, and thereby TORC1 activity. Active TORC1 then releases the Lst4-Lst7 complex from the vacuole to attenuate GAP function of the Lst4-Lst7 complex, thus providing a negative feedback mechanism to tightly control TORC1 activity77.
What and where is the amino acid sensor?
This is a long-standing question in the TOR field. What properties should an amino acid sensor have? First, it should bind directly to amino acids at a physiologically relevant concentration. Second, amino acid binding should affect its function. Third, the sensor should transmit the signal of amino acid availability to TORC1. Sensors have been proposed to exist in the lysosome, mitochondria, and cytoplasm. Zoncu et al.47 suggested that amino acids are obligatorily sensed in the lumen of the lysosome and signal to the RAGs in a V-ATPase-dependent manner, a so-called “inside-out” mechanism. Amino acids promote mTORC1 binding to RAGs on isolated lysosomes in vitro, and inhibition of the V-ATPase prevents mTORC1 binding. In addition, pumping amino acids out of the lysosomal lumen by overexpression of the proton/amino acid transporter 1 (PAT1) prevents amino acid-induced binding of mTORC1 to isolated lysosomes47. According to their lysosome-centric “inside-out” model, Zoncu et al. suggested that amino acids, including leucine, must accumulate in the lysosomal lumen to activate mTORC1.
How are amino acids transported into the lysosomal lumen and sensed? A recent study demonstrated that leucine flux into the lysosomal lumen is mediated by solute carrier (SLC) family proteins SLC7A5-SLC3A278 that were previously shown to be plasma membrane antiporters that transport leucine into the cell in exchange for glutamine79 (Figure 1). This recent study also found that overexpression of lysosomal-associated transmembrane protein 4b (LAPTM4b) promotes lysosomal localization of SLC7A5-SLC3A2 and thus leucine flux into the lysosome.
Three other independent studies suggested that another SLC family member found in the lysosomal membrane, SLC38A9, is an amino acid sensor for mTORC180,81,82. Biochemical analysis revealed that the transmembrane protein SLC38A9 is part of a RAG-RAGULATOR supercomplex. All three studies showed that SLC38A9 is necessary for amino acids to activate mTORC1 in a RAG- and RAGULATOR-dependent manner. However, V-ATPase is dispensable for SLC38A9-mediated activation of mTORC1, suggesting that SLC38A9 and V-ATPase act in parallel upstream of the RAG-RAGULATOR complex. The interaction between SLC38A9 and the RAG-RAGULATOR complex is strengthened upon amino acid starvation, consistent with the observation that SLC38A9 preferentially binds the inactive form of RAG proteins. Furthermore, in vitro amino acid transport assays using liposomes reconstituted with SLC38A9 oriented with its N terminus facing out, equivalent to its orientation in the lysosomal membrane, suggest that SLC38A9 transports glutamine and arginine, but not leucine, into the lysosome80,81. Rebsamen et al.81 further showed that SLC38A9 pumps glutamine out of liposomes with a rate greater than its glutamine influx rate. However, the transport rate of SLC38A9 is relatively low compared with other amino acid transporters. Interestingly, overexpression of an N-terminal cytoplasmic tail of SLC38A9 (minimum amino acids 1-112) causes mTORC1 to be insensitive to amino acid starvation, indicating that the cytoplasmic tail of SLC38A9 is sufficient to activate mTORC1. This gain-of-function phenotype conferred by a soluble cytoplasmic tail also suggests that amino acid transport activity is not required for mTORC1 activation. Altogether, these studies propose that SLC38A9 is a lysosomal amino acid sensor upstream of mTORC1. Consistent with the in vitro data, arginine, but not leucine, failed to activate mTORC1 in the absence of SLC38A9, suggesting that SLC38A9 is an arginine sensor80. It is still unclear whether arginine is sensed in the lysosomal lumen or the cytoplasm. Although SLC38A9 also transports glutamine, the ability of SLC38A9 to signal glutamine availability to mTORC1 in cells has not been tested. The observation that overexpression of a dominant-negative RAG heterodimer blocks mTORC1 activation by SLC38A9 overexpression suggests that SLC38A9 acts upstream of RAG80. Curiously, Rebsamen et al.81 and Jung et al.82 found that mTORC1 remains bound to the lysosome in SLC38A9 knockdown cells, suggesting that SLC38A9 is not required for RAG-mediated mTORC1 recruitment to the lysosome. SLC38A9 homologs in yeast comprise seven vacuolar amino acid transporters (Avts). Of these Avt proteins, Avt1, 2, and 3 contain an N-terminal region similar to the N terminus of SLC38A9. It would be of interest to determine whether the yeast homologs of SLC38A9 play an evolutionarily conserved role in TORC1 signaling. Yet another lysosomal solute carrier protein, SLC15A4, has been linked to mTORC1 activity in B cells83. SLC15A4-knockout B cells accumulate histidine in the lysosome and display increased lysosomal pH, decreased V-ATPase activity, and reduced mTORC1 activity. Although this study showed that SLC15A4 and mTORC1 colocalize to the lysosome, the role of SLC15A4 in mTORC1 signaling remains to be clarified.
In addition to or contrary to the lysosome-centric “inside-out” model of amino acid sensing, recent evidence suggests that amino acids, especially leucine and glutamine, are sensed outside the lysosome. Han et al.84 reported that the cytoplasmic protein leucyl-tRNA synthetase (LRS) senses leucine to promote mTORC1 lysosomal localization and activity. This study further showed LRS acts as a GAP specifically for RAGD. However, the role of LRS as a GAP has been questioned75. Tsun et al.75 reported that LRS does not have GAP activity. The reason for this discrepancy is not known. Cdc60, the yeast ortholog of LRS, physically interacts with Gtr1 and is necessary and sufficient to activate TORC1 in response to leucine stimulation85 (Figure 2). Genetic analysis also suggests that Cdc60 acts upstream of Gtr1. The studies of Han et al.84 and Bonfils et al.85 are consistent with the notion that LRS/Cdc60 is a cytoplasmic leucine sensor upstream of TORC1. Interestingly, Wolfson et al.86 very recently demonstrated that leucine binds the cytoplasmic protein SESN2 and thereby inhibits SESN2-GATOR2 interaction. Thus, SESN2 is almost certainly a cytoplasmic leucine sensor86, although it remains to be shown whether leucine-mediated inhibition of the SENS2-GATOR2 interaction impinges on GATOR1 and RAGs.
Duran et al.87,88 suggested that glutaminolysis, the double deamination of glutamine catalyzed by glutaminase (GLS) and glutamate dehydrogenase (GDH) to produce α-ketoglutarate (αKG), activates RAG-mTORC1 through prolyl hydroxylase (PHD). αKG is a co-factor for PHD. Leucine binds and activates GDH, the enzyme that catalyzes the second deamination step in glutaminolysis, leading to αKG production. Since glutaminolysis takes place in mitochondria, this mechanism is distinct from lysosomal sensing (Figure 1). The glutaminolysis model provides a sensing mechanism for the activation of mTORC1 by leucine and glutamine.
RAG-independent activation of mTORC1 in response to nutrients
Unlike deletion of TORC1 components, deletion of GTR1 and GTR2 is not lethal in yeast. In mammals, although whole-body RAGA knockout in mice is embryonic lethal89, RAGA-knockout MEFs and RAGA- and B-deficient cardiomyocytes sustain mTORC1 activity (see below)90. These observations hint at the existence of alternative or compensatory mechanism of TORC1 activation independent of RAG/Gtr. Stracka et al.91 recently showed that glutamine, a nitrogen source and the major nutrient input for TORC1 in yeast, activates TORC1 in the absence Gtr1 or Vam6. Consistent with this yeast study, glutamine promotes lysosomal translocation and activation of mTORC1 in RAGA- and RAGB-deficient mammalian cells92, suggesting that RAG/Gtr-independent activation of TORC1 by glutamine may be an evolutionarily conserved mechanism. In addition, Jewell et al.92 demonstrated that RAGULATOR, but not V-ATPase, is dispensable for glutamine-mediated mTORC1 lysosomal translocation and activation. In this case, the small GTPase ADP-ribosylation factor 1 (ARF1) appears to substitute for RAG in mTORC1 activation (Figure 1). The molecular mechanism by which ARF1 senses glutamine and controls mTORC1 requires further investigation.
RAG-independent activation of mTORC1 was further demonstrated by Thomas et al.93. This study identified Ypt1 in yeast and RAB1A in mammals as Gtr- and RAG-independent, respectively, regulators of TORC1. Mechanistically, amino acids promote GTP loading of RAB1A, which in turn stimulates RHEB-mTORC1 interaction on the Golgi (Figure 1). How and where amino acid availability is transduced to RAB1A need further investigation.
Amino acid sensing branch of mTORC1 signaling in mice
Efeyan et al.66 generated and characterized RAGA-Q66L knock-in mice in which RAGA is preferentially loaded with GTP (RAGA-GTP mice). Unlike TSC1- or TSC2-knockout mice which die at embryonic day 9.5–13.515,94,95, RAGA-GTP mice exhibit normal embryonic development and are born with the expected Mendelian ratio66. However, these mice are unable to survive beyond postnatal day 1. Autophagy supplies amino acids for hepatic gluconeogenesis to prevent hypoglycemia in newborn mice. Constitutive activation of mTORC1 in RAGA-GTP mice inhibits autophagy and thereby prevents supply of gluconeogenic amino acids in the liver. This leads to hypoglycemia, faster breakdown of hepatic glycogen, energetic collapse, and ultimately neonatal death. Similar neonatal lethality was observed in mice with hyperactive mTORC1 due to deletion of all three SESNs59. These studies suggest that neonatal inhibition of RAG-mTORC1 signaling and subsequent activation of autophagy are critical for survival.
Although RAGA-GTP mice show no abnormalities during embryonic development, RAGA knockout mice display developmental aberrations and die at embryonic day 10.589. Compared with RAGA, RAGB is less expressed in most organs, and is dispensable for normal development and mTORC1 activity at least in brain and liver. RAGB might compensate for embryonic loss of RAGA since RAGB expression is mildly upregulated in RAGA-knockout mice, and RAGA and RAGB double knockout embryos die slightly earlier than RAGA-knockout embryos. Indeed, liver-specific RAGA loss is partially compensated by upregulation of RAGB expression, and loss of RAGB in RAGA-deficient hepatocytes further reduces mTORC1 signaling. Liver-specific RAGA-knockout mice show reduced mTORC1 activity and, most likely due to loss of the negative feedback loop, higher AKT activity. Higher AKT activity is reflected in increased inhibitory phosphorylation of the transcription factor FOXO and decreased FOXO-mediated transcription of the phosphoenolpyruvate carboxykinase (PEPCK), glucose-6-phosphatase (G6P), and glucokinase genes. Reduced expression of these gluconeogenic genes may explain the mild reduction in blood glucose levels observed in the liver-specific RAGA-knockout mice. Like knockout in the embryo, acute knockout of RAGA in adult mice is lethal. Within 2-3 weeks after induction of RAGA knockout, mice die due to intestinal atrophy. Deletion of RAGA in adult mice also expands monocytes in the bone marrow and spleen. The relationship among lethality, intestinal atrophy, and monocytosis remains to be determined.
Kim et al.90 generated cardiomyocyte-specific RAGA and RAGB double knockout mice, which exhibited cardiac hypertrophy and cardiomyopathy, leading to heart dysfunction. Unlike whole-body knockout89, single knockout of RAGA or RAGB in cardiomyocytes did not confer a heart defect, suggesting that RAGA and RAGB have functionally redundant roles in cardiomyocytes. Surprisingly, mTORC1 activity was not impaired in the cardiomyocyte-specific RAGA/B double knockout mice. The authors speculate that upregulated PI3K-Akt signaling might compensate for loss of RAG in the activation of mTORC190.
Unlike cultured cells, cells in their natural setting never experience complete depletion of amino acids. This raises the question of whether amino acid-induced lysosomal translocation of mTORC1 happens in vivo. Recently, Chen et al.96 demonstrated that acute amino acid infusion promotes mTORC1 lysosomal translocation and activation in the kidney. Moreover, chronic amino acid infusion increases protein synthesis and kidney size, presumably due to hyperactivation of mTORC1. It would be of particular interest to investigate whether amino acid-induced mTORC1 translocation to lysosomes in vivo is dependent on RAGs, RAGULATOR, or V-ATPase, and whether mTORC1 lysosomal translocation happens in other tissues such as liver and muscle where mTORC1 plays a critical role in whole-body metabolism6,97,98,99,100,101,102,103,104.
Human diseases associated with defects in amino acid sensing
In contrast to well-characterized tumorigenic mutations in PI3KCA, TSC1, and TSC2 that affect the growth factor signaling pathway, mutations affecting amino acid sensing were recognized only recently. Before identification of RAGULATOR as an upstream regulator of mTORC1, RAGULATOR component LAMTOR2, originally known as p14, was linked to a primary human immunodeficiency syndrome characterized by short stature, hypopigmented skin, coarse facial features and recurrent bronchopulmonary infection by Streptococcus pneumoniae105. Genetic linkage studies and genome-wide transcriptional profiling analyses of the human primary immunodeficiency syndrome identified a homozygous point mutation in the 3′ untranslated region (UTR) of LAMTOR2/p14. It was then shown that the 3′UTR mutation indeed reduces stability of the mRNA and thus decreases protein expression. LAMTOR2/p14 was previously characterized as a positive regulator of a mitogen-activated protein kinase (MAPK) pathway106. It is not clear whether the disease is due to reduced MAPK or mTORC1 activity. However, cells derived from patients have reduced mTORC1 activity38, and this defect appears to be more severe than the defect in MAPK signaling105. Since TORC1 controls cell and organism size, and rapamycin is immunosuppressive, it seems likely that reduced mTORC1 activity accounts for the disease phenotype.
Mutation of NPRL2 or DEPDC5, genes encoding GATOR1 components, has been linked to lung, breast, ovarian, kidney, liver and brain cancers50,107,108,109,110,111,112,113. Indeed, cancer cells harboring homozygous loss of function mutations in GATOR1 components have elevated mTORC1 activity that is insensitive to amino acid starvation50. Introduction of a functional GATOR1 component in these cells restored mTORC1 sensitivity to amino acid depletion and reduced cell proliferation. These findings suggest that GATOR1 is a tumor suppressor complex upstream of mTORC1. Two independent exome-sequencing analyses of familial focal epilepsy identified loss-of-function mutations in DEPDC5114,115, suggesting that hyperactive mTORC1 may cause epilepsy and mTORC1 inhibitors may be used to treat this disorder.
Birt-Hogg-Dube (BHD) syndrome is a rare disease characterized by multiple benign skin and hair follicle tumors. Patients with BHD syndrome also have increased risk of developing cysts in the lung and high incidence of kidney tumors. Loss-of-function mutations in the FLCN gene cause the BHD syndrome, and FLCN is thus considered a tumor suppressor116. Indeed, kidney-specific knockout of FLCN causes upregulation of MAPK and PI3K-Akt-mTOR signaling, which may explain the kidney hyperplasia observed in FLCN-knockout mice72,117. The renal hyperplasia was at least in part due to upregulation of mTORC1 signaling since rapamycin treatment reduced kidney size and extended lifespan. Consistent with the mouse study, renal tumors from BHD patients also showed upregulation of mTORC1 activity72,117. However, it is counterintuitive that FLCN, a positive regulator of mTORC1, is a tumor suppressor. Future studies are required to clarify the apparent discrepancy that FLCN behaves like an oncogene in cultured cells (see section on FLCN above) but is a tumor suppressor in the BHD syndrome.
RAB1A overexpression is frequently observed in tongue squamous carcinomas, colorectal cancer, and hepatocellular carcinoma, and correlates with hyperactive mTORC1 signaling and poor prognosis93,118. Cell proliferation assays in vitro and xenograft studies in vivo further demonstrated that RAB1A overexpression promotes oncogenic growth of cancer cells in a rapamycin-sensitive manner.
Closing remarks and future directions
The finding that RAG proteins regulate TORC1 in response to nutrients is a milestone in TOR research. Follow-up studies to elucidate regulators of the RAG proteins have expanded the list of molecules involved in amino acid sensing, and studies in mice have provided important insight into the physiology and pathophysiology of amino acid sensing. However, several questions remain.
How are amino acids sensed?
The answer to this question is more complex than previously expected. Increasing evidence suggests that different amino acids are sensed at distinct locations in the cell and signal to mTORC1 in RAG-dependent and -independent manners. Investigation of upstrem regulator of the FNIP-FLCN complex may lead to yet unknown amino acid sensors. For RAG-independent sensing, studying upstream regulators of ARF1 may provide a glutamine sensor. It would also be of interest to examine whether glutamine signals to mTORC1 via a common sensor in RAG-dependent and -independent manners. Intriguingly, loss of TORC1 function in yeast is lethal whereas deletion of both GTR1 and GTR2 (gtr1Δ gtr2Δ cells) is not lethal119. Furthermore, glutamine signals to TORC1 in a Gtr1-independent manner. Thus, a yeast synthetic lethal screen in grt1Δgtr2Δ cells might identify a mechanism by which cells sense and transduce glutamine availability to TORC1.
How important is amino acid sensing in the whole animal?
Studies on amino acid signaling in mice have provided unexpected insight. For example, acute deletion of RAGA in adult mice causes monocytosis89, revealing a previously unrecognized role of mTORC1 in this disease characterized, curiously, by monocyte proliferation. Follow-up studies using cell type-specific RAGA-knockout mice are needed to elucidate the role of amino acid signaling in monocyte function and presumably immunity.
Although amino acid infusion promotes mTORC1 lysosomal translocation in the kidney, further studies are required to demonstrate the extent of this form of regulation in the whole animal. As discussed above, amino acids are rarely depleted in the animal. What is the physiological amino acid concentration that elicits lysosomal translocation of mTORC1 and how often is this achieved? To this end, amino acid levels in vivo should be examined by metabolomic analysis and correlated with mTORC1 lysosomal translocation. Experiments such as amino acid infusion in cell type-specific RAG-knockout mice will indicate whether findings in vitro apply in vivo.
Recent findings revealed that the amino acid signaling pathway is clinically relevant. Mutations in pathway components are linked to human diseases. Furthermore, cancer cells harboring mutations in components of GATOR1 or overexpressing RAB1A are hypersensitive to mTORC1 inhibition50,93. Thus, alterations in the amino acid signaling pathway could serve as biomarkers to predict efficacy of mTORC1 inhibitors. Further analysis of mice carrying mutations in disease-causing genes such as NPRL2 or DEPDC5 will provide more insight into the role of amino acid sensing in disease. Finally, examination of patients in which mTORC1 activity is deregulated may provide more information on amino acid sensing in human physiology.
References
Wullschleger S, Loewith R, Hall MN . TOR signaling in growth and metabolism. Cell 2006; 124:471–484.
Laplante M, Sabatini DM . mTOR signaling in growth control and disease. Cell 2012; 149:274–293.
Dibble CC, Manning BD . Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 2013; 15:555–564.
Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD . A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans 2013; 41:906–912.
Lamming DW, Sabatini DM . A Central role for mTOR in lipid homeostasis. Cell Metab 2013; 18:465–469.
Albert V, Hall MN . mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol 2015; 33:55–66.
Shimobayashi M, Hall MN . Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 2014; 15:155–162.
Efeyan A, Comb WC, Sabatini DM . Nutrient-sensing mechanisms and pathways. Nature 2015; 517:302–310.
Cornu M, Albert V, Hall MN . mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev 2013; 23:53–62.
Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 110:163–175.
Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 110:177–189.
Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 2002; 10:457–468.
Gao X, Zhang Y, Arrazola P, et al. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol 2002; 4:699–704.
Kenerson HL, Aicher LD, True LD, Yeung RS . Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res 2002; 62:5645–5650.
Kwiatkowski DJ, Zhang H, Bandura JL, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet 2002; 11:525–534.
Onda H, Crino PB, Zhang H, et al. Tsc2 null murine neuroepithelial cells are a model for human tuber giant cells, and show activation of an mTOR pathway. Mol Cell Neurosci 2002; 21:561–574.
Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J . Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA 2002; 99:13571–13576.
Dibble CC, Elis W, Menon S, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell 2012; 47:535–546.
Pearce LR, Komander D, Alessi DR . The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010; 11:9–22.
Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 1996; 15:6541–6551.
Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997; 7:261–269.
Inoki K, Li Y, Zhu T, Wu J, Guan KL . TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002; 4:648–657.
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC . Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 2002; 10:151–162.
Menon S, Dibble CC, Talbott G, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014; 156:771–785.
Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J . Rheb binds and regulates the mTOR kinase. Curr Biol 2005; 15:702–713.
Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006; 126:955–968.
Um SH, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004; 431:200–205.
Hsu PP, Kang SA, Rameseder J, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011; 332:1317–1322.
Yu Y, Yoon SO, Poulogiannis G, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011; 332:1322–1326.
Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J . Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem 1998; 273:14484–14494.
Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C . The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell 2005; 19:15–26.
Powis K, Zhang T, Panchaud N, Wang R, Virgilio CD, Ding J . Crystal structure of the Ego1-Ego2-Ego3 complex and its role in promoting Rag GTPase-dependent TORC1 signaling. Cell Res 2015; 25:1043–1059.
Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL . Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008; 10:935–945.
Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320:1496–1501.
Schurmann A, Brauers A, Massmann S, Becker W, Joost HG . Cloning of a novel family of mammalian GTP-binding proteins (RagA, RagBs, RagB1) with remote similarity to the Ras-related GTPases. J Biol Chem 1995; 270:28982–28988.
Hirose E, Nakashima N, Sekiguchi T, Nishimoto T . RagA is a functional homologue of S. cerevisiae Gtr1p involved in the Ran/Gsp1-GTPase pathway. J Cell Sci 1998; 111:11–21.
Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T . Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J Biol Chem 2001; 276:7246–7257.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM . Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141:290–303.
Binda M, Peli-Gulli MP, Bonfils G, et al. The Vam6 GEF controls TORC1 by activating the EGO complex. Mol Cell 2009; 35:563–573.
Nakashima N, Noguchi E, Nishimoto T . Saccharomyces cerevisiae putative G protein, Gtr1p, which forms complexes with itself and a novel protein designated as Gtr2p, negatively regulates the Ran/Gsp1p G protein cycle through Gtr2p. Genetics 1999; 152:853–867.
Valbuena N, Guan KL, Moreno S . The Vam6 and Gtr1-Gtr2 pathway activates TORC1 in response to amino acids in fission yeast. J Cell Sci 2012; 125:1920–1928.
Demetriades C, Doumpas N, Teleman AA . Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014; 156:786–799.
Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM . Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012; 150:1196–1208.
Nada S, Hondo A, Kasai A, et al. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J 2009; 28:477–489.
Kogan K, Spear ED, Kaiser CA, Fass D . Structural conservation of components in the amino acid sensing branch of the TOR pathway in yeast and mammals. J Mol Biol 2010; 402:388–398.
Zhang T, Peli-Gulli MP, Yang H, De Virgilio C, Ding J . Ego3 functions as a homodimer to mediate the interaction between Gtr1-Gtr2 and Ego1 in the ego complex to activate TORC1. Structure 2012; 20:2151–2160.
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM . mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011; 334:678–683.
Forgac M . Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 2007; 8:917–929.
Dechant R, Saad S, Ibanez AJ, Peter M . Cytosolic pH regulates cell growth through distinct GTPases, Arf1 and Gtr1, to promote Ras/PKA and TORC1 activity. Mol Cell 2014; 55:409–421.
Bar-Peled L, Chantranupong L, Cherniack AD, et al. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013; 340:1100–1106.
Panchaud N, Peli-Gulli MP, De Virgilio C . Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci Signal 2013; 6:ra42.
Neklesa TK, Davis RW . A genome-wide screen for regulators of TORC1 in response to amino acid starvation reveals a conserved Npr2/3 complex. PLoS Genet 2009; 5:e1000515.
Dokudovskaya S, Waharte F, Schlessinger A, et al. A conserved coatomer-related complex containing Sec13 and Seh1 dynamically associates with the vacuole in Saccharomyces cerevisiae. Mol Cell Proteomics 2011; 10:M110.006478.
Panchaud N, Peli-Gulli MP, De Virgilio C . SEACing the GAP that nEGOCiates TORC1 activation: evolutionary conservation of Rag GTPase regulation. Cell Cycle 2013; 12:2948–2952.
Dokudovskaya S, Rout MP . SEA you later alli-GATOR — a dynamic regulator of the TORC1 stress response pathway. J Cell Sci 2015; 128:2219–2228.
Lee JH, Budanov AV, Karin M . Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab 2013; 18:792–801.
Budanov AV, Karin M . p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008; 134:451–460.
Lee JH, Budanov AV, Park EJ, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010; 327:1223–1228.
Peng M, Yin N, Li MO . Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 2014; 159:122–133.
Chantranupong L, Wolfson RL, Orozco JM, et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep 2014; 9:1–8.
Parmigiani A, Nourbakhsh A, Ding B, et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep 2014; 9:1281–1291.
Kim JS, Ro SH, Kim M, et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci Rep 2015; 5:9502.
Jin G, Lee SW, Zhang X, et al. Skp2-mediated RagA ubiquitination elicits a negative feedback to prevent amino-acid-dependent mTORC1 hyperactivation by recruiting GATOR1. Mol Cell 2015; 58:989–1000.
Deng L, Jiang C, Chen L, et al. The ubiquitination of RagA GTPase by RNF152 negatively regulates mTORC1 activation. Mol Cell 2015; 58:804–818.
Nakayama K, Nagahama H, Minamishima YA, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 2000; 19:2069–2081.
Efeyan A, Zoncu R, Chang S, et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 2012; 493:679–683.
van Slegtenhorst M, Khabibullin D, Hartman TR, Nicolas E, Kruger WD, Henske EP . The Birt-Hogg-Dube and tuberous sclerosis complex homologs have opposing roles in amino acid homeostasis in Schizosaccharomyces pombe. J Biol Chem 2007; 282:24583–24590.
Chan TF, Carvalho J, Riles L, Zheng XF . A chemical genomics approach toward understanding the global functions of the target of rapamycin protein (TOR). Proc Natl Acad Sci USA 2000; 97:13227–13232.
Liu W, Chen Z, Ma Y, Wu X, Jin Y, Hou S . Genetic characterization of the Drosophila birt-hogg-dube syndrome gene. PLoS One 2013; 8:e65869.
Hartman TR, Nicolas E, Klein-Szanto A, et al. The role of the Birt-Hogg-Dube protein in mTOR activation and renal tumorigenesis. Oncogene 2009; 28:1594–1604.
Hudon V, Sabourin S, Dydensborg AB, et al. Renal tumour suppressor function of the Birt-Hogg-Dube syndrome gene product folliculin. J Med Genet 2010; 47:182–189.
Baba M, Furihata M, Hong SB, et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst 2008; 100:140–154.
Takagi Y, Kobayashi T, Shiono M, et al. Interaction of folliculin (Birt-Hogg-Dube gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene 2008; 27:5339–5347.
Petit CS, Roczniak-Ferguson A, Ferguson SM . Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J Cell Biol 2013; 202:1107–1122.
Tsun ZY, Bar-Peled L, Chantranupong L, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell 2013; 52:495–505.
Roberg KJ, Bickel S, Rowley N, Kaiser CA . Control of amino acid permease sorting in the late secretory pathway of Saccharomyces cerevisiae by SEC13, LST4, LST7 and LST8. Genetics 1997; 147:1569–1584.
Peli-Gulli MP, Sardu A, Panchaud N, Raucci S, De Virgilio C . Amino acids stimulate TORC1 through Lst4-Lst7, a GTPase-activating protein complex for the Rag Family GTPase Gtr2. Cell Rep 2015; 13:1–7.
Milkereit R, Persaud A, Vanoaica L, Guetg A, Verrey F, Rotin D . LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat Commun 2015; 6:7250.
Nicklin P, Bergman P, Zhang B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009; 136:521–534.
Wang S, Tsun ZY, Wolfson RL, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015; 347:188–194.
Rebsamen M, Pochini L, Stasyk T, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015; 519:477–481.
Jung J, Genau HM, Behrends C . Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol Cell Biol 2015; 35:2479–2494.
Kobayashi T, Shimabukuro-Demoto S, Yoshida-Sugitani R, et al. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity 2014; 41:375–388.
Han JM, Jeong SJ, Park MC, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012; 149:410–424.
Bonfils G, Jaquenoud M, Bontron S, Ostrowicz C, Ungermann C, De Virgilio C . Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol Cell 2012; 46:105–110.
Wolfson RL, Chantranupong L, Saxton RA, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2015 Oct 8. pii: aab2674
Duran RV, Mackenzie ED, Boulahbel H, et al. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene 2012; 32:4549–4556.
Duran RV, Oppliger W, Robitaille AM, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 2012; 47:349–358.
Efeyan A, Schweitzer LD, Bilate AM, et al. RagA, but not RagB, is essential for embryonic development and adult mice. Dev Cell 2014; 29:321–329.
Kim YC, Park HW, Sciarretta S, et al. Rag GTPases are cardioprotective by regulating lysosomal function. Nat Commun 2014; 5:4241.
Stracka D, Jozefczuk S, Rudroff F, Sauer U, Hall MN . Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J Biol Chem 2014; 289:25010–25020.
Jewell JL, Kim YC, Russell RC, et al. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015; 347:194–198.
Thomas JD, Zhang YJ, Wei YH, et al. Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell 2014; 26:754–769.
Kobayashi T, Minowa O, Sugitani Y, et al. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc Natl Acad Sci USA 2001; 98:8762–8767.
Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ . Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest 1999; 104:687–695.
Chen JK, Nagai K, Chen J, et al. Phosphatidylinositol 3-kinase signaling determines kidney size. J Clin Invest 2015; 125:2429–2444.
Bentzinger CF, Romanino K, Cloetta D, et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab 2008; 8:411–424.
Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM . mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010; 468:1100–1104.
Kenerson HL, Yeh MM, Yeung RS . Tuberous sclerosis complex-1 deficiency attenuates diet-induced hepatic lipid accumulation. PLoS One 2011; 6:e18075.
Yecies JL, Zhang HH, Menon S, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 2011; 14:21–32.
Bentzinger CF, Lin S, Romanino K, et al. Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skelet Muscle 2013; 3:6.
Castets P, Lin S, Rion N, et al. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab 2013; 17:731–744.
Cornu M, Oppliger W, Albert V, et al. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc Natl Acad Sci USA 2014; 111:11592–11599.
Umemura A, Park EJ, Taniguchi K, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab 2014; 20:133–144.
Bohn G, Allroth A, Brandes G, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med 2007; 13:38–45.
Teis D, Wunderlich W, Huber LA . Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev Cell 2002; 3:803–814.
Lerman MI, Minna JD . The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer Res 2000; 60:6116–6133.
Zabarovsky ER, Lerman MI, Minna JD . Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene 2002; 21:6915–6935.
Li J, Wang F, Haraldson K, et al. Functional characterization of the candidate tumor suppressor gene NPRL2/G21 located in 3p21.3C. Cancer Res 2004; 64:6438–6443.
Seng TJ, Ichimura K, Liu L, Tingby O, Pearson DM, Collins VP . Complex chromosome 22 rearrangements in astrocytic tumors identified using microsatellite and chromosome 22 tile path array analysis. Genes Chromosomes Cancer 2005; 43:181–193.
Ueda K, Kawashima H, Ohtani S, et al. The 3p21.3 tumor suppressor NPRL2 plays an important role in cisplatin-induced resistance in human non-small-cell lung cancer cells. Cancer Res 2006; 66:9682–9690.
Miki D, Ochi H, Hayes CN, et al. Variation in the DEPDC5 locus is associated with progression to hepatocellular carcinoma in chronic hepatitis C virus carriers. Nat Genet 2011; 43:797–800.
Yogurtcu B, Hatemi I, Aydin I, Buyru N . NPRL2 gene expression in the progression of colon tumors. Genet Mol Res 2012; 11:4810–4816.
Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013; 45:552–555.
Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013; 45:546–551.
Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2002; 2:157–164.
Hasumi Y, Baba M, Ajima R, et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci USA 2009; 106:18722–18727.
Xu BH, Li XX, Yang Y, et al. Aberrant amino acid signaling promotes growth and metastasis of hepatocellular carcinomas through Rab1A-dependent activation of mTORC1 by Rab1A. Oncotarget 2015; 6:20813–20828.
Gao M, Kaiser CA . A conserved GTPase-containing complex is required for intracellular sorting of the general amino-acid permease in yeast. Nat Cell Biol 2006; 8:657–667.
Acknowledgements
We acknowledge support from the Louis-Jeantet Foundation, the Swiss National Science Foundation and the Canton of Basel.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shimobayashi, M., Hall, M. Multiple amino acid sensing inputs to mTORC1. Cell Res 26, 7–20 (2016). https://doi.org/10.1038/cr.2015.146
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2015.146
Keywords
This article is cited by
-
Female developmental environment delays development of male honeybee (Apis mellifera)
BMC Genomics (2021)
-
Plasma concentrations of branched-chain amino acids differ with Holstein genetic strain in pasture-based dairy systems
Scientific Reports (2021)
-
A glutamine sensor that directly activates TORC1
Communications Biology (2021)
-
Progression and metastasis of small cell lung carcinoma: the role of the PI3K/Akt/mTOR pathway and metabolic alterations
Cancer and Metastasis Reviews (2021)
-
Limited survival and impaired hepatic fasting metabolism in mice with constitutive Rag GTPase signaling
Nature Communications (2021)
