
Abstract
Brain ischemia often results in neuronal necrosis, which may spread death to neighboring cells. However, the molecular events of neuronal necrosis and the mechanisms of this spreading death are poorly understood due to the limited genetic tools available for deciphering complicated responses in mammalian brains. Here, we engineered a Drosophila model of necrosis in a sub-population of neurons by expressing a leaky cation channel in the Drosophila eye. Expression of this channel caused necrosis in defined neurons as well as extensive spreading of cell death. Jun N-terminal kinase (JNK)-mediated, caspase-independent apoptosis was the primary mechanism of cell death in neurons, while caspase-dependent apoptosis was primarily involved in non-neuronal cell death. Furthermore, the JNK activation in surrounding neurons was triggered by reactive oxygen species (ROS) and Eiger (Drosophila tumor necrosis factor α (TNFα)) released from necrotic neurons. Because the Eiger/ROS/JNK signaling was also required for cell death induced by hypoxia and oxidative stress, our fly model of spreading death may be similar to brain ischemia in mammals. We performed large-scale genetic screens to search for novel genes functioning in necrosis and/or spreading death, from which we identified several classes of genes. Among them, Rho-associated kinase (ROCK) had been reported as a promising drug target for stroke treatment with undefined mechanisms. Our data indicate that ROCK and the related trafficking pathway genes regulate neuronal necrosis. We propose the suppression of the function of the trafficking system, ROS and cytokines, such as TNFα, as translational applications targeting necrosis and spreading death.
Similar content being viewed by others

Main
As the second leading cause of death worldwide, brain ischemia often results in necrotic cell death in the infarct core and cellular dysfunction in the peri-infarct zone known as ischemic penumbra.1 Because dysfunctional cells in the penumbra may be rescued through prompt reperfusion within a few hours after the insult, penumbral salvage has been the major aim of therapy.2, 3 However, most patients are admitted to clinics many hours after the insult, resulting in permanent damage in the penumbra. In such cases, alleviation of cell death and inflammation may be more critical for recovery.4 Indeed, some patients suffered from progressive dementia even years after the insult.2 Currently, no effective neuroprotective therapy has been developed.2, 5 One reason is that our limited knowledge of cell death restricts the design of treatment strategies.5
In ischemia and reperfusion, extremely complicated cellular and molecular events induce acute and progressive cell death, including excitotoxicity, energy loss, Jun N-terminal kinase (JNK) activation, generation of reactive oxygen species (ROS), release of apoptosis-inducing factor (AIF) and cytokines (such as tumor necrosis factor α (TNFα)), metabolic changes, protein aggregation, organelle damage and synaptic failure.6, 7, 8, 9 Sometimes, the causal or responsive roles of these changes on cell death are hard to dissect. Many questions remain, such as how adjacent cells respond to the necrotic infarct core, what death signals are released to spread death, and whether there are genetic pathways regulating cell death.
Here, we built a genetic system to induce necrosis in a sub-population of neurons in Drosophila, and we studied its effect on neighboring cells. To induce neuronal necrosis, a leaky cation channel was expressed to overload the cells with calcium. Our data indicate that neuronal necrosis results in extensive spreading of death to adjacent cells through complicated signaling pathways, including ROS, JNK, Eiger (Drosophila TNFα) and other unknown signals. Furthermore, these spreading death signals are also required for cell death induced by hypoxia and oxidative stress, suggesting that they are relevant to ischemic stroke in mammals. Moreover, we performed large-scale genetic screens and identified several classes of proteins that may regulate primary necrosis and/or spreading death. Our model provides a genetic tool to dissect the molecular mechanisms of necrosis and spreading death, which may mimic the molecular events in the ischemic brain.
Results
Expressing a leaky cation channel in a subset of neurons in the Drosophila eye caused extensive cell death and eye defects
To induce neuronal necrosis, we generated a transgenic fly line to express a constitutively open cation channel, the mouse glutamate receptor 1 Lurcher mutant (GluR1Lc).10 Then, UAS-GluR1Lc was expressed in fly eyes, driven by the sevenless-Gal4 (sev-Gal4) promoter. The fly progenies from sev-Gal4 and UAS-GluR1Lc are simplified as sev>GluR1Lc (the binary UAS/Gal4 expression system is represented as ‘>’ throughout the text). The compound eyes of Drosophila are formed by nearly 800 units of small eyes, known as ommatidia, each of which contains 8 photoreceptor cells (or R cells).11 During development in the larval eye disc, R8 recruits the R2/R5 pair and the R3/R4 pair, and they form a five-cell pre-cluster. In the adult stage, the R1/R6 pair and R7 are also recruited into the ommatidium.11 The sev-Gal4 promoter is specifically expressed in the R3/R4 pair of the larval eye disc and R3/R4/R7 of the adult eye.12 In the GluR1Lc-expressing larvae, we verified that the transgenic GluR1Lc protein was located in the plasma membrane (Supplementary Figure S1A) and that calcium overloading by GluR1Lc indeed occurred in Drosophila neurons (Supplementary Figure S1B).
In sev>GluR1Lc flies, the adult eye size was greatly reduced (Figures 1Aa and b), as were the numbers of ommatidia and bristles (Figures 1Ac–d1). Strikingly, few cells were identifiable in the cross-sectioned ommatidia (Figures 1Ae and f). By transmission electron microscopy (TEM), the damaged cells exhibited loss of plasma membrane integrity and emergence of intracellular vacuoles (Figures 1Ag and h). These results suggest that massive death occurred in neuronal and non-neuronal cells in the adult eyes. At the larval stage, the GFP fluorescent intensity in the eye disc of the sev>GluR1Lc/eGFP (UAS-eGFP could visualize the GluR1Lc-expressing cells) flies was greatly reduced (Figure 1B), suggesting that cell death occurred in the third instar larval stage, after the sev-Gal4 promoter began to express.

Characterization of necrosis induced by sev>GluR1Lc. (A) Effect of GluR1Lc expression. (a and b) Light images. (c and d) SEM images. (c1 and d1) Enlarged images from (c) and (d), respectively. (e and f) Sectioned adult eyes stained with toluidine blue. (g and h) Images from TEM. (B) Confocal images of larval eye discs (a) sev-Gal4 driven UAS-GFP to show sev expression pattern; (b) sev-Gal4 driven UAS-GFP and UAS-GluR1Lc to show increased cell death. (C) Effects of caspase inhibitors on the eye defect of sev>GluR1Lc flies. (a–d) The controls showed that UAS-P35, UAS-IAP1 and UAS-IAP2 blocked apoptosis (GMR-hid), as expected. (e–h) These anti-apoptotic lines had no effect on the sev>GluR1Lc eye defect. (D) Immunostaining with anti-cleaved-caspase 3 to detect caspase activity. As a positive control, cleaved caspase-3 activity was detected in the GMR-hid flies (a), but not in the sev>GluR1Lc larval eye disc (b). (E) Staining with PI to detect necrosis. Anti-GFP and anti-GluR1 label the sev-expressing cells. DAPI labels nuclei. PI signal was undetectable in the eye disc of wild-type flies (a) or apoptotic flies (b). However, PI and anti-GluR1 were colocalized in the sev>GluR1Lc flies, suggesting that GluR1Lc-expressing cells died from necrosis (c). (F) ROS level change detected by DHE staining in larval eye discs (a) sev>GFP the control; (b) sev>rpr/GFP -Gal4 induced apoptosis in the sev-expressing cells; (c) the sev>GluR1Lc model. (G) LysoTracker staining. Many GluR1Lc-expressing neurons could be stained with LysoTracker, indicating that these cells became acidic. (a) sev>GFP the control; (b–d) the sev>GluR1Lc model stained by anti-GFP (b), Lyso-tracker (c), and merged image from b and c (d)
Occurrence of neuronal necrosis in GluR1Lc-expressing neurons
The death of GluR1Lc-expressing neurons was not mediated by caspase-mediated apoptosis because caspase inhibition had no effect on the eye defect (Figure 1C). Consistently, caspase activity could not be detected by anti-cleaved-caspase immunostaining in the larval eye disc (Figure 1D). In contrast, there was positive staining for the necrotic marker propidium iodide (PI) in the GluR1Lc-expressing neurons (Figure 1E). In addition, we examined other features of necrosis, including mitochondrial defects, elevation of ROS level and increase in cytoplasmic acidification,13 all of which took place in the GluR1Lc-expressing neurons (Supplementary Figure S1C; Figures 1F and G). Together, these results suggest that the GluR1Lc-expressing neurons die from necrosis.
Spreading apoptotic death in adjacent cells induced by primary neuronal necrosis
Because the sev-Gal4 promoter drives GluR1Lc expression in two of the five R cells in larvae and three of the eight R cells in adult in each ommatidium, the other R cells should stay alive. However, the remaining number of neurons was far lower than expected (Figure 1Af), suggesting the occurrence of spreading death. One caveat is that spreading death may be mediated through gap junctions because the R cells can form gap junctions during development.14 We think this scenario is unlikely because only GluR1Lc-expressing cells undergo necrosis. In addition, we found that mutants of several gap junction proteins did not affect fly eye size ablation (Supplementary Table S1).
To determine whether the additional deaths were from neurons or glial cells, the larval eye discs were stained with a neuronal marker (22C10, which stains the cytoskeleton) and a glial marker (Repo, which stains the nucleus). We found that spreading death mainly occurs in the posterior region of eye discs, which contains mostly mature neurons (Figures 2Aa–d). To further confirm primary (GluR1Lc-expressing neurons) or spreading (non-GluR1Lc-expressing) death, we co-stained the samples with anti-GluR1 and anti-ELAV (which stains neuronal nuclei) (Figures 2Ba–c1). We observed that the newly formed ommatidia in the anterior region of sev>GluR1Lc eye discs were relatively normal (Figures 2Bd and f1). However, in the posterior region, the ELAV staining was diminished in the GluR1-positive R3/4 cells (Figure 2Bf2), and it became clumpy in the adjacent neurons (Figures 2Bf2 and f3). These results clearly show that spreading death occurs in adjacent neurons at the larval stage.

Spreading cell death from primary necrotic neurons. (A) Staining by a neuronal (22C10) and a glial cell marker (Repo) in the larval eye disc. 22C10 staining was decreased (a and b) in the eye disc of sev>GluR1Lc flies, but Repo showed no change (c and d). (B) Morphological change of neurons in larval eye disc. Neurons were labeled by anti-ELAV, and sev-expressing neurons were labeled by anti-GFP in wild type (sev>mCD8-GFP) (a–c1) or anti-GluR1 in sev>GluR1Lc flies (d–f3). In sev>GluR1Lc, ELAV staining was reduced (d–f). Enlarged individual ommatidia showed progressive defects of R3/4 pair (GluR1+) and R2/5/8(GluR1−) in sev>GluR1Lc (f1–f3). (C) Immunostaining with anti-GFP and anti-ELAV to show that no spreading death occurred in the eye disc of sev>rpr/mCD8-GFP flies (a–c2). (D) Image of the adult eye sev>rpr under light microscope (a), SEM (b and b1) and sectioned adult eye stained by toluidine blue (c)
In addition, neuronal apoptosis induced by sev>rpr could not spread death in the eye disc (Figures 2C and D). Therefore, only necrosis could spread death.
Caspase-dependent spreading apoptosis in non-neuronal cells
Although caspase activation was minimal in the third instar eye disc (Figure 1D), we found that inhibition of caspases in all ommatidial cells by GMR-P35 strongly rescued the adult eye size defect (Figures 3Aa and b). Similar effects were also observed in the double mutants of effector caspases Dcp-1 and drICE (Figure 3Ac), and in a deficiency line (H99) that simultaneously removes the three inhibitor of apoptosis protein (IAP) antagonists, hid, rpr and grim (Figure 3Ad). Further analysis using hidWR+X1, rpr87 or either one in combination with H99 (Figures 3Ae–h) suggests that the caspase-dependent apoptosis is mediated by hid. To determine which cell type was rescued by GMR-P35, we sectioned adult eyes. Non-neuronal cells, but not R cells, were strongly rescued (compare the numbers of white vacuoles in Figures 3Bb and c). We quantified the R cell salvage by ‘ommatidia density,’ the total number of identifiable ommatidia in a fixed area (1000 μm2). This quantification showed that the ommatidia density in wild type, sev>GluR1Lc and GMR-P35; sev>GluR1Lc flies was 7.63, 0.81 and 1.34, respectively (Figure 3Bd). This result suggests that caspase-dependent apoptosis accounts for ∼8% (1.34–0.81/7.63–0.81) of spreading death in neurons. We exclude the possibility that one copy of GMR-P35 might be too weak to inhibit caspase activity (Supplementary Figure S2A). Furthermore, we observed that cell death markers, including acridine orange (AO) and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL), could positively label many cells in the larval eye discs, and their staining patterns were unaltered by GMR-P35 (Figure 3C). This suggests that extensive apoptosis is caspase independent. We further verified that TUNEL staining mainly labels the apoptotic cells and not the necrotic cells (co-staining of TUNEL with PI or GluR1Lc-expressing neurons, Supplementary Figure S2B).

Characterization of spreading death in sev>GluR1Lc. (A) Representative light images of sev>GluR1Lc in different apoptotic mutants (a–h). We measured the eye size (as area) with Adobe Photoshop and normalized the ratio to sev>GluR1Lc. Lines that rescue, enhance or have no effect are represented by green, red or black letters, respectively, throughout later figures. The genotypes tested are listed in Supplementary Information. (B) Toluidine blue staining of sectioned adult eye. GMR-P35 showed limited rescue of R cells (a–c). (d) Quantitative analysis of the ommatidia density. The number of ommatidia in a fixed area (1000 μm2) was counted. Bars represent the mean±standard deviation throughout the later figures. Trial n=5. Asterisks represent statistical significance for t-test or one-way ANOVA tests for multi-comparison, ***P<0.001, **P<0.01, *P<0.05. N.S. denotes no significance in statistics in this Figure, and throughout the later Figures. (C) TUNEL and AO staining in the larval eye disc. sev>GluR1Lc flies had significant increased AO (a–c) and TUNEL staining (d–f)
JNK-mediated caspase-independent spreading apoptosis in neurons
To study how caspase-independent apoptosis is activated, we examined two pathways that are reported to not respond to the caspase inhibitor P35 in Drosophila, the JNK and AIF pathways.15, 16 JNK signaling requires a cascade of protein kinases, including JNK, JNK Kinase (JNKK) and JNKK Kinase (JNKKK).17 We found that hep1/Y, hepr75/+ (JNKK mutants), bsk1/+ (a JNK mutant) and TRAF2Ex (a mutant in a component upstream of JNKKK) all rescued the eye defects of sev>GluR1Lc (Figures 4Aa–f). In contrast, AIFKO/+ (AIF knockout) showed no effect (Figure 4Ag). In addition, the pucE69 allele, a JNK gain-of-function (GOF) mutant,18 enhanced the eye defect (Figure 4Ah). However, expressing bskDN (dominant negative) or hepRNAi (Figures 4Ai and j) in GluR1Lc-expressing neurons had no effect, indicating that JNK signaling acts in adjacent cells. We further confirmed that JNK-mediated death was caspase independent by testing the effect of bsk1 in the presence of GMR-P35, which showed additional rescue compared with GMR-P35 alone (Figures 4Ak and m). Consistently, pucE69 could further enhance the GMR-P35;sev>GluR1Lc phenotype (Figure 4Al). Quantification of the ommatidia density suggests that ∼47% of spreading apoptosis in R cells can be rescued by the hepr75 and bsk1 double mutants (Figure 4Bd), suggesting that JNK-mediated apoptosis is the major form of spreading death in neurons. Moreover, JNK signaling was indeed activated in the larval stage (Figures 4Ca and c) and further elevated at the pupal stage, as determined by a JNK reporter in vivo, puc-lacZ19 (Figures 4Cb and d). Because anti-β-Gal was partly colocalized with anti-ELAV (Figures 4Ce and e1) but not with anti-GluR1 (Figure 4Cf), we conclude that JNK activation only occurred in adjacent neurons.

JNK signaling is required for the non-caspase spreading death. (A) Representative light images adult eye of sev>GluR1Lc under different mutant backgrounds. (a–j) Under sev>GluR1Lc background. (k–m) Under GMR-P35/+; sev>GluR1Lc/+; background. (B) Toluidine blue staining (a–c). (d) Quantification of rescue effect by ommatidia density. Trail n=3. (e) Ratio of different types of death pathways. (C) JNK activation in the larval and pupal eye disc. (a and c) The third instar larval eye disc. (b and d) Pupal stage at 60 h after pupa formation (a.p.f.). Also note that the pupal eye layer of sev>GluR1Lc is thinner than wild-type flies. (e and e1) Different channels of the same sample co-stained with anti-β Gal and anti-ELAV. (f) Anti-β-Gal co-staining with anti-GluR1
JNK-mediated apoptosis was triggered by the release of Eiger and ROS from adjacent neurons undergoing primary necrosis
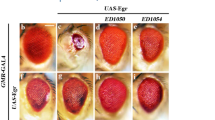
Previous studies suggest that JNK-mediated death can be activated by eiger expression driven by a pan-eye promoter GMR-Gal4 (GMR>eiger) in Drosophila.20 Strikingly, when driven by the sev-Gal4 promoter, UAS-eiger (sev>eiger) failed to elevate the puc-lacZ intensity or generate an eye defect (Figure 5Ae). Actually, the lacZ intensity was slightly reduced (see also later text), indicating that eiger expression was not sufficient for JNK activation. However, overexpression of eiger in necrotic neurons (sev>GluR1Lc/eiger) enhanced the puc-lacZ intensity and eye size defect (Figure 5Ad). Knocking down eiger in necrotic neurons with eigerRNAi (sev>GluR1Lc/ eigerRNAi) significantly diminished the elevated puc-lacZ level and rescued the eye size defect (Figures 5Aa–c) without affecting the primary necrosis (Supplementary Figure S3Ac and d). Together, these results suggest that eiger from necrotic neurons serves as a spreading factor to activate JNK signal in the adjacent cells.

Eiger and ROS are required for JNK activation in adjacent neurons. (A) Eiger is required for JNK activation. (a–c) eigerRNAi reduced the adult eye defect and larval LacZ staining in sev>GluR1Lc. (d) UAS-eiger enhanced eye defects and LacZ staining. (e) sev>eiger failed to induce eye defects and puc-LacZ staining. (f) GMR>eiger is a positive control. (B) Increasing ROS enhanced JNK activity. (a and b) ROS level (detected by DHE staining) was increased in the eye disc of sev>GluR1Lc/ND75RNAi flies. (c and d) JNK activity was further increased in sev>GluR1Lc/ND75RNAi flies. (C) Chelating ROS reduced JNK activity. (a–c) Expressing catalase or catalase and GTPx-1 rescued the eye size defect. (d) JNK activation was strongly reduced by expression of catalase and GTPx-1 in sev>GluR1Lc
Stroke-induced oxidative stress is well known to cause neuronal death.21 We found that expressing ND75RNAi (inhibiting mitochondrial complex I to increase ROS production)22 in necrotic neurons enhanced ROS production (determined by dihydroethidium (DHE) staining, Figures 5Ba and b) and activated JNK signaling (Figures 5Bc and d). Functionally, overexpression of catalase (ROS chelating enzyme) or the combination of catalase with GTPx-1 (another ROS chelating enzyme) reduced the eye defect (Figures 5Ca–c) and JNK activation (Figure 5Cd) without affecting primary necrosis (Supplementary Figures S3b and d). This suggests that ROS from necrotic cells is also required for JNK-mediated apoptosis.
Intracellular ROS is required for cell-autonomous apoptosis but not for spreading apoptosis
A previous report suggested that intracellular ROS levels are elevated in the GMR>eiger flies, possibly inducing a necroptosis-like death in Drosophila photoreceptor neurons.20 In addition, the JNK-mediated cell death might be apoptotic or necrotic depending on cell types and stimuli.23, 24 Our results demonstrate that Eiger-mediated death in R cells is caused by apoptosis, based on negative PI staining (Supplementary Figure S3B) and mutants of necroptosis components, including loss-of-function (LOF) PGMAM5 and Drp125 (Supplementary Figure S3C). Next, we examined the functional role of ROS in spreading death, and found that chelating ROS by overexpression of catalase or GTPx-I strongly suppressed Eiger-induced death (Supplementary Figures S3Da–e). Therefore, intracellular ROS is a key effector of Eiger-mediated death. In contrast, intracellular ROS in the apoptotic cells could not further enhance the JNK signal (Supplementary Figure S3E). This result is consistent with our earlier data (Figures 2C and D). Therefore, secondary apoptosis around the primary necrotic core may slow down the spreading necrotic insults (Figure 6F).

Eiger/ROS/JNK signaling in oxidative stress. (A) Eiger/ROS/JNK signaling is required for cell death induced by 0.03% H2O2 in larval eye disc. The OA staining patterns were shown in a–d. Their bright field images were shown in the lower panel (a1–d1). (a) H2O2 could induce cell death, as determined by AO staining. (b–d) The cell death induced by H2O2 could be suppressed by eigerRNAi or overexpression of catalase, GTPx-1 or hep1 mutant. The ratio of positive AO spots in a fixed area (every confocal 63 × 3 field, 6684.04 μm2) relative to the control is shown in the right panel. (B) Eiger is required for JNK activation by 0.03% H2O2 in larval eye disc. (a and b) Ectopically applied H2O2 activates JNK signaling. (c) H2O2 treatment could not activate JNK signaling (indicated by lacZ staining) in the eye disc of GMR>eigerRNAi flies. (C) Real-time PCR for eiger transcripts. Trial n=3. The result showed that H2O2 treatment activated eiger transcripts. (D) Immunostaining by anti-Eiger in the larval eye disc of w1118 flies. The result shows that the protein level of Eiger is higher after H2O2 treatment. (E) Immunostaining by anti-Eiger and anti-GFP. The data showed that H2O2 treatment disrupted plasma membrane of the cells and more Eiger distributed into the extracellular space. (F) Model of spreading death in the sev>GluR1Lc flies. Primary necrotic neurons release ROS and Eiger to spread death. The ratio of different spreading apoptosis is shown. For JNK-mediated death, extracellular ROS triggers expression and release of Eiger, which activates JNK and intracellular ROS. Intracellular ROS cannot further activate JNK signaling and therefore terminates spreading death by apoptosis
Requirement of the Eiger/ROS/JNK signaling in cell death induced by hypoxia or oxidative stress
In addition to calcium overloading, neuronal death can be triggered by hypoxia and oxidative stress during ischemic stroke in mammals.26 Therefore, we ask whether the Eiger/ROS/JNK signaling is also required for cell death-induced by hypoxia and oxidative stress in Drosophila. To induce hypoxia-mediated death, the third instar larva was immersed in the Drosophila hemolymph HL3 saline.27 Twenty hours after hypoxia, ROS levels were greatly elevated in the eye discs (Supplementary Figures S4Aa). The result showed that overexpression of catalase and GTPx-1 or LOF of Eiger and JNK significantly diminished the ROS elevation (Supplementary Figures S4Ab–d). For oxidative stress, we treated the larval eye discs with ectopically applied H2O2 (0.003%, g/ml) in tissue culture. After 10 h of treatment, AO staining was significantly increased (Figure 6Aa). Functionally, GOF constructs for catalase and GTPx-1 (GMR>catalase/GTPx-1), LOF eiger (GMR>eiger RNAi) or hep (hep1) all rescued the cell death (Figures 6Ab–e). Together, these results suggest that the Eiger/ROS/JNK signaling pathway is required for hypoxia and oxidative stress-induced cell death in Drosophila.
The role of extracellular ROS in eiger expression and release
Reducing JNK signaling could rescue the cell death in the Drosophila eye disc under H2O2 stress. Consistently, we also found JNK activation when treating eye discs expressing a JNK reporter, puc-lacZ, with H2O2 (Figures 6Ba and b). Interestingly, GMR>eigerRNAi could suppress the JNK activation, suggesting that eiger works downstream of H2O2 (Figure 6Bc). Mechanistically, H2O2 treatment could increase transcript and protein levels of Eiger (Figures 6C and D). In addition, H2O2 may enhance Eiger release (Figure 6E). Taken together, extracellular ROS may activate JNK through Eiger by promoting the latter’s transcription and release.
In addition to the activation of JNK, we observed that overexpression of Eiger in the sev>eiger flies could suppress the JNK signal at basal conditions or after H2O2 treatment (Supplementary Figure S4). These results indicate that low levels of Eiger GOF may antagonize JNK signaling. Consistently, TNFα pretreatment has been shown to be neuroprotective in the mammalian brain after stroke, likely through activation of ceramide, a second messenger involved in multiple functions.28
Genetic screen for modifiers of sev>GluR1Lc
To identify dominant suppressors for sev>GluR1Lc, we performed a genome-wide screen using a deficiency kit. We found 23 strong suppressors from nearly 400 deficiency lines that cover most of the genome (Table 1). As positive controls, three deficiency lines disrupting the IAP antagonist and JNKK were identified. Previously, mutations in the metabolic energy production pathways have been identified as suppressing Eiger-induced death in Drosophila.20 Consistently, we identified five deficiency lines (three metabolic mutants and two undefined lines) that may be shared with that screen (Table 1).20
We also performed a GOF screen using ∼3400 EPgy2 lines29 and an LOF screen using nearly 1000 TRiP RNAi lines.30 From these screens, we identified 15 strong suppressors (9 for EPgy2 and 6 for RNAi). Together, seven functional groups were identified, including genes functioning in caspase-dependent apoptosis, JNK signaling and metabolism, oxidation-reduction, trafficking, ubiquitination, nuclear export and synapse activity (Table 1). Because the EPgy2 and RNAi lines are based on the UAS/Gal4 system, the modifiers from these two screens should mainly affect primary necrosis. In theory, they may also affect the spreading factors, such as Eiger or ROS.
Opposing effects of metabolic pathways on primary necrosis and spreading apoptosis
For the class 2 genes, we observed apparently contradictory results. The two metabolic point mutations (wal02516 and cyt-c-dbln−1) appeared as suppressors, while the RNAi lines appeared as enhancers (Figures 7Aa–g; Table 1). Because their effects were unaltered in the GMR-P35;sev>GluR1Lc flies, these genes should not affect caspase-dependent apoptosis (Figures 7Aa1–g1). We found that these genes have opposing effects on primary necrosis and Eiger-mediated apoptosis; their LOF suppressed Eiger-mediated apoptosis (Figure 7B) and enhanced primary necrosis (Figures 7C and D).

Mechanism study of modifiers from the genetic screen. (A) Effects of metabolic pathways on the eye defect of sev>GluR1Lc flies. a–g1, Representative images are shown for LOF mutants from the Deficiency screen (some are point mutations) and TRiP RNAi screens on sev>GluR1Lc and GMR-P35;sev>GluR1Lc eye size. (B) Effect of the metabolic pathway mutants in (A) on GMR>eiger eye size (a–g). (C) PI staining of the eye discs of sev>GluR1Lc flies under some of the metabolic pathway mutants and bsk1 background (a–h). (D) Quantification of PI staining in a fixed area (1000 μm2) of the third instar larval eye disc. All the metabolic mutants enhanced the PI staining. Mutation of the upstream gene bsk, bsk1, did not affect necrosis. Three eye discs were examined for each line. (E) Characterization of representative suppressors in the class 4 modifiers from the genetic screen. ROCKRNAi, sqhRNAi and GOF β’ cop all rescue the adult eye size (a–d) and PI staining (e–h). (e1–g1) Co-staining of anti-GluR1 and PI. The genotypes are the same as in (a–c). (l) Quantitative data of PI staining. Primary necrosis was reduced in these lines. Three eye discs were examined for each line
Regulation of neuronal necrosis and spreading death by diverse family of proteins
From these screens, gene functions in trafficking (class 4) were enriched, including three genes in the small GTPase pathway (Rho1, Rho-associated kinase (ROCK) and sqh) and two genes in another small GTPase pathway (rab8 and rab9) (Figures 7Ea–d). As a key regulator of cytoskeleton and cell polarity, the small GTPase pathway has been suggested to be involved in a wide range of diseases, including stroke and Alzheimer’s disease.31, 32 We verified that ROCKRNAi and sqhRNAi indeed rescued primary necrosis in the larval eye disc of the sev>GluR1Lc flies (Figure 7Ee–g and e1–g1). In addition, GOF β’Cop, another trafficking gene,33 rescued primary necrosis (Figure 7Eh).
We also identified genes functioning in ubiquitination, nuclear transport and synaptic structure (Table 1, class 5–7). For the ubiquitin pathway, we identified cul-2, cul-3, Ubi-p63E and Cct5. These genes may involve in proteasomal function,34 protein degradation35 and neuromuscular junction development.36 We also identified three genes related to synaptic activities, including lbm, Nsf2 and wnt2 (Table 1). These genes are required for synapse assembly,37 synaptic transmission or protein localization in the neuromuscular junction.38
From these screens, we conclude that neuronal necrosis and spreading death involve distinct gene functions and cellular events.
Discussion
Our model provides a genetic system of neuronal necrosis and its spreading death in the Drosophila eye. More broadly, this model may improve our understanding of the complicated molecular and cellular events in stroke, especially primary necrosis, necrosis-mediated spreading death, and interactions between necrotic neurons and their adjacent cells.
Furthermore, our screen had identified proteins that function in multiple pathways, such as the ubiquitin proteasomal pathway, nuclear export and synaptic formation, all of which have been implicated in ischemic stroke.39, 40 In general, neuronal necrosis has been considered as an irreversible process because of the severe organelle damage.41 Our screening result suggests that this damage process may involve multiple distinct genetic pathways. Therefore, it is possible to target necrotic progression.
For the mechanism of spreading death, our data demonstrate that different cell types may use different strategies in response to spreading insults. For instance, activation of JNK in neurons may trigger caspase-independent apoptosis, likely through Eiger. Activation of hid in non-neuronal cells may promote caspase-dependent apoptosis. It is possible that the hid pathway is also mediated by Eiger.42 Moreover, other unknown death pathways may also trigger neuronal apoptosis. Further, using our simplified genetic model, we found that manipulating ROS and Eiger in GluR1Lc-expressing neurons could affect the JNK-dependent death and the eye defect without affecting the primary necrosis. Therefore, the spreading death is initiated by ROS and Eiger from the primary necrotic neurons.
Necrotic cells can induce apoptosis by releasing toxic factors. We propose that apoptosis may terminate the necrotic insults for the following reasons. First, caspase-dependent apoptosis induced by sev>rpr could not spread death. Second, TNFα/JNK/ROS-mediated death could not further spread death because ROS generated in the JNK-dependent apoptosis could not further activate JNK signaling. Regarding TNFα/JNK/ROS signaling, some researchers have proposed that a positive feedback loop from TNF to JNK and then to ROS may enhance cell death because elevated ROS can activate JNK signal43, 44 and associate with TNFα and JNK-mediated death.24 Our data indicate that such a feedback loop does not exist during necrosis-mediated spreading death in Drosophila.
Regarding the translational applications of cell protection, our data suggest targeting several pathways, including neuronal necrosis, caspase-dependent and -independent death-spreading pathways, and likely other undefined pathways. Our data suggest that chelating ROS may be beneficial. Interestingly, edaravone, an ROS scavenger drug, has been tested in clinical trials for the treatment of ischemic stroke.45 Targeting TNFα may also be desirable because it is critical for triggering JNK-mediated apoptosis. Recently, targeting TNFα with an engineered chimeric monoclonal antibody that could pass the blood–brain barrier has been shown to be neuroprotective in a mouse model of stroke.46 Another advantage of targeting TNFα is that reducing its level may activate its preconditioning function. Our genetic modeling is limited by the fact that necrosis induced by the leaky channel may not reflect physiological conditions in human disease. Further investigations are required to determine whether the proteins identified here are suitable drug targets.
Materials and Methods
Fly maintenance and stocks
Drosophila stocks were raised on standard sucrose/cornmeal medium at constant 25°C with a 12-h light/dark cycle. The following Drosophila strains are kind gifts from various laboratories: pucE69-lacZ and UAS-eigerRNAi (Dr. Tian Xu); AIFKO (Dr. Josef Penninger); UAS-IAP2 (Dr. Pascal Meier); UAS-cat II and UAS-GTPx-I III (Dr. Utpal Banerjee); hep1/FM, TRAF2Ex/FM and UAS-eiger (Dr. Lei Xue); GMR-hid, GMR-Gal4, Dcp-1Prev/Dcp-1Prev, drICEDelta1/Tb, H99/Tb, hidWR+X1 and rpr87 (Dr. Yun Fan and Dr. Andreas Bergmann). TRiP RNAi stocks were obtained from the Tsinghua Drosophila stock center. Other lines were obtained from the Bloomington Stock Center.
Generation of transgenic fly lines
UAS-GluR1Lc transgenic Drosophila was generated in a w1118 background using the pUAST vector. The mGluR1Lc fragment was digested from pCAGGS-mGluR1Lc construct (a gift from Dr. Michisuke Yuzaki) and subcloned into the pUAST vector.
Histology in adult eyes
For section preparation, 3- to 5-day-old adult heads were fixed, and the eyes were sectioned at 1 μm using an ultramicrotome (UC7; Leica, Tokyo, Japan). Then, the slides were stained with toluidine blue for 30 s at 80°C. For TEM, the eye samples were sectioned at 200–400 nm. The later procedures followed a standard protocol.
Histology of larval tissue staining
AO, TUNEL, immunostaining, DHE, TMRM and lacZ staining were performed as described.22, 47, 48, 49, 50 For the PI (P4170; Sigma, St Louis, MO, USA) staining, the freshly dissected eye or brain complexes were incubated with 10 μg/ml PI solution (in Schneider’s medium) for 10 min at room temperature. Antibodies used include anti-GFP (A11122; Invitrogen, Eugene, OR, USA), anti-cleaved caspase 3 (9661; Cell Signaling, Danvers, MA, USA), anti-GluR1 (ab 32132; Abcam, Eugene, OR, USA), chicken anti-β-Gal (ab9361; Abcam, Eugene, OR, USA), mouse anti-β-Gal (40-1a; DSHB, Iowa city, IA, USA), anti-ELAV (9F8A9; DSHB), anti-22C10 (22C10; DSHB) and anti-Repo (8D12; DSHB). Anti-Eiger was a gift from Dr. Masayuki Miura.
Calcium imaging by Fura-2
The early third instar larvae of ElavGS>GluR1Lc or ElavGS-Gal4>eGFP (as a control) were incubated with 5 mg/ml RU486 (mifepristone M8046; Sigma) for 90 s.51 After 5 h of induction, we dissected the larval brains and incubated them with 5 μM Fura-2 AM (F-1221; Invitrogen Molecular Probe, Eugene, OR, USA) in Schneider’s medium at 25°C for 30 min in the dark. We recorded two emission intensity values at 510 nm, with excitation wavelengths at 340 and 380 nm, respectively. The ratio of emission intensity at 510 nm excited by 340 or 380 nm was calculated as the relative calcium concentration.
Drosophila hypoxia assay
The third instar larvae were incubated in a 1.5-ml tube with HL3 saline for 45 min. Then, the larvae were returned to normal vials containing regular food. Normally, these larvae recovered within 15 min. However, at 10–15 h after hypoxia, these larvae started to die. At 20 h after hypoxia, we dissected the eye disc and stained them with DHE.
Abbreviations
- GluR1Lc:
-
glutamate receptor 1 Lurcher mutant
- sev:
-
sevenless
- TNFα:
-
tumor necrosis factor α
- ROS:
-
reactive oxygen species
- JNK:
-
Jun N-terminal kinase
- PI:
-
presidium iodide
- AO:
-
acridine orange
- DHE:
-
dihydroethidium
- AIF:
-
apoptosis-inducing factor
- IAP:
-
inhibitor of apoptosis protein
- JNKK:
-
JNK Kinase
- JNKKK:
-
JNKK Kinase
- TEM:
-
transmission electron microscopy
- ROCK:
-
Rho-associated kinase
- GOF:
-
gain-of-function
- LOF:
-
loss-of-function
- a.p.f.:
-
after pupa formation
References
Astrup J, Symon L, Branston NM, Lassen NA . Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke 1977; 8: 51–57.
Liu R, Yuan H, Yuan F, Yang SH . Neuroprotection targeting ischemic penumbra and beyond for the treatment of ischemic stroke. Neurol Res 2012; 34: 331–337.
Lo EH . A new penumbra: transitioning from injury into repair after stroke. Nat Med 2008; 14: 497–500.
Dirnagl U, Iadecola C, Moskowitz MA . Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999; 22: 391–397.
Yuan J . Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis 2009; 14: 469–477.
Zheng Z, Lee JE, Yenari MA . Stroke: molecular mechanisms and potential targets for treatment. Curr Mol Med 2003; 3: 361–372.
Hofmeijer J, van Putten MJ . Ischemic cerebral damage: an appraisal of synaptic failure. Stroke 2011; 43: 607–615.
Wojcik C, Di Napoli M . Ubiquitin-proteasome system and proteasome inhibition: new strategies in stroke therapy. Stroke 2004; 35: 1506–1518.
Hayashi T, Abe K . Ischemic neuronal cell death and organellae damage. Neurol Res 2004; 26: 827–834.
Kohda K, Wang Y, Yuzaki M . Mutation of a glutamate receptor motif reveals its role in gating and delta2 receptor channel properties. Nat Neurosci 2000; 3: 315–322.
Tomlinson A, Ready DF . Neuronal differentiation in Drosophila ommatidium. Dev Biol 1987; 120: 366–376.
Tomlinson A, Bowtell DD, Hafen E, Rubin GM . Localization of the sevenless protein, a putative receptor for positional information, in the eye imaginal disc of Drosophila. Cell 1987; 51: 143–150.
Syntichaki P, Samara C, Tavernarakis N . The vacuolar H+ -ATPase mediates intracellular acidification required for neurodegeneration in C. elegans. Curr Biol 2005; 15: 1249–1254.
Shimohigashi M, Meinertzhagen IA . The shaking B gene in Drosophila regulates the number of gap junctions between photoreceptor terminals in the lamina. J Neurobiol 1998; 35: 105–117.
Joza N, Galindo K, Pospisilik JA, Benit P, Rangachari M, Kanitz EE et al. The molecular archaeology of a mitochondrial death effector: AIF in Drosophila. Cell Death Differ 2008; 15: 1009–1018.
Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J 2002; 21: 3009–3018.
Stronach BE, Perrimon N . Stress signaling in Drosophila. Oncogene 1999; 18: 6172–6182.
Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 1998; 12: 557–570.
Adachi-Yamada T, Fujimura-Kamada K, Nishida Y, Matsumoto K . Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature 1999; 400: 166–169.
Kanda H, Igaki T, Okano H, Miura M . Conserved metabolic energy production pathways govern Eiger/TNF-induced nonapoptotic cell death. Proc Natl Acad Sci USA 2011; 108: 18977–18982.
Niizuma K, Endo H, Chan PH . Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem 2009; 109 (Suppl 1): 133–138.
Owusu-Ansah E, Yavari A, Mandal S, Banerjee U . Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet 2008; 40: 356–361.
Moreno E, Yan M, Basler K . Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 2002; 12: 1263–1268.
Ventura JJ, Cogswell P, Flavell RA, Baldwin AS Jr., Davis RJ . JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev 2004; 18: 2905–2915.
Wang Z, Jiang H, Chen S, Du F, Wang X . The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012; 148: 228–243.
Moskowitz MA, Lo EH, Iadecola C . The science of stroke: mechanisms in search of treatments. Neuron 2010; 67: 181–198.
Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF . Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A 1994; 175: 179–191.
Ginis I, Schweizer U, Brenner M, Liu J, Azzam N, Spatz M et al. TNF-alpha pretreatment prevents subsequent activation of cultured brain cells with TNF-alpha and hypoxia via ceramide. Am J Physiol 1999; 276 (5 Pt 1): C1171–C1183.
Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 2004; 167: 761–781.
Ni JQ, Zhou R, Czech B, Liu LP, Holderbaum L, Yang-Zhou D et al. A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods 2011; 8: 405–407.
Amano M, Nakayama M, Kaibuchi K . Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010; 67: 545–554.
Mueller BK, Mack H, Teusch N . Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov 2005; 4: 387–398.
Kreis TE, Lowe M, Pepperkok R . COPs regulating membrane traffic. Annu Rev Cell Dev Biol 1995; 11: 677–706.
Lee HS, Simon JA, Lis JT . Structure and expression of ubiquitin genes of Drosophila melanogaster. Mol Cell Biol 1988; 8: 4727–4735.
Ou CY, Pi H, Chien CT . Control of protein degradation by E3 ubiquitin ligases in Drosophila eye development. Trends Genet 2003; 19: 382–389.
Ayyub C . Cullin-5 and cullin-2 play a role in the development of neuromuscular junction and the female germ line of Drosophila. J Genet 2011; 90: 239–249.
Kopczynski CC, Davis GW, Goodman CS . A neural tetraspanin, encoded by late bloomer, that facilitates synapse formation. Science 1996; 271: 1867–1870.
Liebl FL, McKeown C, Yao Y, Hing HK . Mutations in Wnt2 alter presynaptic motor neuron morphology and presynaptic protein localization at the Drosophila neuromuscular junction. PLoS One 2012; 5: e12778.
Hu BR, Martone ME, Jones YZ, Liu CL . Protein aggregation after transient cerebral ischemia. J Neurosci 2000; 20: 3191–3199.
Hochrainer K, Jackman K, Anrather J, Iadecola C . Reperfusion rather than ischemia drives the formation of ubiquitin aggregates after middle cerebral artery occlusion. Stroke 2012; 43: 2229–2235.
Taoufik E, Probert L . Ischemic neuronal damage. Curr Pharm Des 2008; 14: 3565–3573.
Ma X, Huang J, Yang L, Yang Y, Li W, Xue L . NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res 2011; 22: 425–431.
Owusu-Ansah E, Banerjee U . Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009; 461: 537–541.
Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y et al. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J 2003; 22: 3898–3909.
Group EAIS. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc Dis 2003; 15: 222–229.
Sumbria RK, Boado RJ, Pardridge WM . Brain protection from stroke with intravenous TNFalpha decoy receptor-Trojan horse fusion protein. J Cereb Blood Flow Metab 2012; 32: 1933–1938.
Arama E, Steller H . Detection of apoptosis by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling and acridine orange in Drosophila embryos and adult male gonads. Nat Protoc 2006; 1: 1725–1731.
Sweeney ST, Hidalgo A, de Belle JS, Keshishian H . X-gal staining of the central nervous system in adult Drosophila. Cold Spring Harb Protoc 2012; 2012: 239–241.
Wu JS, Luo L . A protocol for dissecting Drosophila melanogaster brains for live imaging or immunostaining. Nat Protoc 2006; 1: 2110–2115.
Chazotte B . Labeling mitochondria with TMRM or TMRE. Cold Spring Harb Protoc 2011; 2011: 895–897.
Osterwalder T, Yoon KS, White BH, Keshishian H . A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci USA 2001; 98: 12596–12601.
Acknowledgements
This work was supported by grants from the Chinese Ministry of Science and Technology (2009CB941300 and 2013CB530700), National Natural Science Foundation of China (NSFC31171324) and Peking-Tsinghua Center for Life Sciences to LL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Yang, Y., Hou, L., Li, Y. et al. Neuronal necrosis and spreading death in a Drosophila genetic model. Cell Death Dis 4, e723 (2013). https://doi.org/10.1038/cddis.2013.232
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.232
Keywords
This article is cited by
-
Vimar/RAP1GDS1 promotes acceleration of brain aging after flies and mice reach middle age
Communications Biology (2023)
-
Amelioration of a neurodevelopmental disorder by carbamazepine in a case having a gain-of-function GRIA3 variant
Human Genetics (2022)
-
Speed Switch in Glioblastoma Growth Rate due to Enhanced Hypoxia-Induced Migration
Bulletin of Mathematical Biology (2020)
-
A Mechanistic Investigation into Ischemia-Driven Distal Recurrence of Glioblastoma
Bulletin of Mathematical Biology (2020)
-
Characterization of TNF-induced cell death in Drosophila reveals caspase- and JNK-dependent necrosis and its role in tumor suppression
Cell Death & Disease (2019)
