
Abstract
The regulation of CCAAT/enhancer-binding protein-homologous protein (CHOP), an endoplasmic reticulum (ER) stress-response factor, is key to cellular survival. Hypoxia is a physiologically important stress that induces cell death in the context of the ER, especially in solid tumors. Although our previous studies have suggested that Cyclophilin B (CypB), a molecular chaperone, has a role in ER stress, currently, there is no direct information supporting its mechanism under hypoxia. Here, we demonstrate for the first time that CypB is associated with p300 E4 ligase, induces ubiquitination and regulates the proteasomal turnover of CHOP, one of the well-known pro-apoptotic molecules under hypoxia. Our findings show that CypB physically interacts with the N-terminal α-helix domain of CHOP under hypoxia and cooperates with p300 to modulate the ubiquitination of CHOP. We also show that CypB is transcriptionally induced through ATF6 under hypoxia. Collectively, these findings demonstrate that CypB prevents hypoxia-induced cell death through modulation of ubiquitin-mediated CHOP protein degradation, suggesting that CypB may have an important role in the tight regulation of CHOP under hypoxia.
Similar content being viewed by others


Main
CCAAT/enhancer-binding protein-homologous protein (CHOP) is thought to have a role in endoplasmic reticulum (ER) stress-induced apoptosis. Furthermore, overexpression of CHOP leads to decrease in the Bcl-2 protein, and overexpression of Bcl-2 blocks CHOP-induced apoptosis.1, 2 It is a leucine-zipper transcription factor that is present at low levels under normal conditions but is highly upregulated in ER stress by ATF4.3, 4, 5, 6 Increased expression of CHOP promotes cell death, but the deletion of the CHOP gene leads to an attenuation in cell death induced by ER stress.2, 7 Also, the accumulation of CHOP protein during ER stress has been reported to be regulated through modulation of its degradation by the proteasome.8 However, recently it has been reported that CHOP is not uniformly essential for cell death induced by ER stress, although CHOP is clearly important in many scenarios of apoptosis.9 Therefore, how CHOP mediates apoptosis has not been clearly understood yet. Recently, we showed that Cyclophilin B (CypB) blocks Ca2+ leakage from the ER to cytosol against the ER stress-inducing drug, thapsigargin (Ca2+ ATPase (SERCA) blocker), and ultimately prevents cell death in response to ER stress.10 However, the exact molecular mechanism by which CypB protects against ER stress is still elusive.
The aim of this study was to explore the possible functional mechanisms underlying CypB in ER stress-induced apoptosis under hypoxia in terms of CHOP. We have shown that hypoxia transcriptionally upregulates CypB through ATF6. CypB interacts with CHOP, one of the best-characterized pro-apoptotic molecules, leading to its p300-mediated ubiquitination and degradation. Subsequently, elevated CypB shows protective effects against hypoxia-induced cell death. In addition, we found that knockdown of CypB could not prevent ER-associated apoptotic signaling under hypoxia. These results suggest that CypB might be another key molecule in the adaptation to hypoxia.
Results
CypB interacts with CHOP and promotes polyubiquitination of CHOP under hypoxia
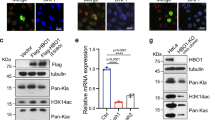
First, we monitored the expression levels of proteins involved in hypoxia-induced cell death in CypB-overexpressing cells under hypoxia. Intriguingly, the expression level of CHOP was significantly decreased. As shown in Figure 1a, the expression level of CHOP protein was decreased by CypB expression in a dose-dependent manner. However, hypoxia still induced CHOP mRNA expression independently of CypB overexpression (Figure 1a, lower panel). These results suggest that CypB may modulate the protein stability of CHOP, but not its transcription level. To investigate in detail whether endogenous CypB contributes to the regulation of CHOP physiologically, we produced AGS cells depleted of endogenous CypB using siRNA for dose-dependent silencing of CypB expression. Importantly, the level of CHOP protein was increased when CypB was silenced under hypoxia, but not with control siRNA (Figure 1b). These results indicate that CypB may regulate the levels of endogenous CHOP.

CypB modulates ubiquitination and degradation of CHOP. (a) AGS cells were transfected with Flag-CypB expression plasmids. After transfection, cells were exposed to hypoxia for 24 h and subjected to western blot (upper panel) or RT-PCR (lower panel). IB, immunoblot. (b) AGS cells transfected with increasing concentrations of CypB siRNA for 48 h were exposed to normoxic or hypoxic conditions for 24 h and then immunoblotted with indicated antibodies. (c) AGS cells were transfected with Flag-CypB (4 μg) expression plasmids and incubated for 4 h in the presence of MG132 (20 μM) and exposed to hypoxic conditions for 24 h. Cell lysates were subjected to western blot. (d) AGS cells transfected with siRNA for either control (Con) or CypB were treated with CHX; 10 μM, and expression of the indicated proteins was determined by immunoblotting at the indicated times. (e) CypB-mediated CHOP ubiquitination. AGS cells were cotransfected with plasmids for HA-ubiquitin (2 μg) and Flag-CypB (4 μg). After transfection, cells were incubated with or without MG132 (20 μM) for 4 h. Cell lysates were immunoblotted with indicated antibodies. (f) siRNA inhibition of CypB reduced CHOP ubiquitination. AGS cells were transfected with siRNA for either Con or CypB, and lysates were immunoblotted with antibodies as indicated in the input panels (left panel). For CHOP ubiquitination, cell lysates was immunoprecipitated with anti-CHOP antibody and immunoblotted with anti-ubiquitin antibody (right panel). Quantitative analysis of all the bands is shown on a bar graph using ImageJ program. Bars represent means±s.d.; *P<0.05, **P<0.01
To gain greater insight into the function of CypB in CHOP regulation, we investigated whether CypB participates in the proteasomal degradation pathway of the CHOP protein. As shown in Figure 1c, overexpression of CypB increased the degradation of hypoxia-induced CHOP protein (Figure 1c, compare lane 2 with lane 5). However, treatment with MG132, an inhibitor of the 26S proteasome system, inhibited CypB-mediated degradation of CHOP protein (Figure 1c, compare lane 5 with lane 6). Moreover, as shown in Figure 1d, the half-life of CHOP protein was longer in CypB siRNA-treated cells compared with control siRNA-treated cells under hypoxia after treatment with cycloheximide (CHX). As CHOP protein is reportedly degraded by ubiquitination,11 we monitored the involvement of CypB in the ubiquitination and degradation of CHOP. AGS cells were transfected with CypB siRNA and CHOP ubiquitination was monitored by western blot. As shown in Figure 1e, lanes 2 and 4, overexpression of CypB facilitated the ubiquitination of endogenous CHOP. To confirm these results, we repeated these experiments with CypB siRNA-treated cells. siRNA-mediated inhibition of CypB led to a significant decrease in ubiquitination and an increase in the stabilization of CHOP (Figure 1f). Together, these results suggest that CypB may modulate CHOP degradation.
Next, we tested for a physical interaction between CHOP and CypB by coimmunoprecipitation in human embryonic kidney (HEK)293 QBI cells. We performed coimmunoprecipitation experiments after inducing the expression of endogenous CypB–CHOP complex under hypoxia in HEK293 QBI cells without exogenous protein expression. The specific CypB–CHOP interaction was checked in immunoprecipitates using anti-CypB. Figure 2a shows the endogenous specific CypB–CHOP complex under hypoxia.

CypB physically interacts with CHOP. (a) Physical interaction of endogenous CypB with endogenous CHOP. AGS cells were exposed to hypoxia for 24 h and incubated for 4 h in the presence of MG132 (20 μM). Equal amounts of AGS cell lysates were immunoprecipitated with negative control IgG and CypB antibodies and immunoblotted with the indicated antibodies. (b and c) Flag-CHOP, WT HA-CypB, and mutant R95A HA-CypB were transfected into HEK293 QBI cells as indicated. Immunoprecipitated products were subjected to western blot using the indicated antibodies to confirm the presence of appropriate proteins. (d) CypB colocalized with CHOP. After AGS cells were transiently transfected with HA-CypB (4 μg) and Flag-CHOP (4 μg) expression plasmids (left panel), or exposed to hypoxia for 24 h (right panel), the cells were incubated in the presence of MG132 (20 μM) for 4 h, followed by an immunocytochemistry experiment. Cells were stained with anti-CypB (green) and anti-CHOP (red). Bars, 10 μm. (e) Schematic representaion of WT CypB (upper panel) and full-length CHOP and the various truncation mutated CHOP constructs (lower panel): ΔN36, α-helix domain deletion; ΔBR, whole-DNA-binding domain deletion; ΔLZ, leucine zipper domain deletion. (f) The α-helix domain of CHOP is critical for CypB binding. HEK293 QBI cells were transiently transfected with the indicated constructs. The cell lysates were immunoprecipitated with anti-Flag antibody, and immunoblotting of cell lysates was performed with anti-Myc or anti-Flag antibodies. Shown is a schematic diagram of CHOP deletion mutants with an indication of the CypB binding domain (lower panel). (g) Detection of the interaction between CypB and CHOP in vitro. Glutathione-agarose beads containing GST or GST-CypB were incubated with whole-cell extracts derived from HEK293 QBI cells expressing WT CHOP, ΔN36, ΔBR, and ΔLZ truncation mutants. (h) HEK293 QBI cells were transiently transfected with an expression vector for WT CypB, WT CHOP, and ΔN36 CHOP. After 24 h, the cell lysates were analyzed by immunoprecipitation using antibodies against the indicated epitope tags. (i) CypB promotes in vivo CHOP ubiquination, but not ΔN36 CHOP. Expressing vectors were transfected with WT CypB, Myc-CHOP truncation mutants and HA-ubiquitin into HEK293 QBI cells as indicated. The cell lysates were immunoprecipitated with anti-Myc antibody, and mutiubiquitinated CHOP were detected by immunoblotting with anti-HA (upper panel). Expression level of each protein was assessed by anti-Myc, anti-Flag, and anti-actin (lower panel)
After cotransfection with HA-CypB and Flag-CHOP constructs, HA antibody was used to immunoprecipitate exogenous HA-CypB, and Flag antibody or HA antibody was used to probe the immunoprecipitates on immunoblots. Flag-CHOP was found to coimmunoprecipitate with HA-CypB in the presence of MG132 (Figure 2b). Intriguingly, we could not show any significantly weaker interaction between Flag-CHOP and PPIase-defective CypB R95A mutant, suggesting that PPIase activity of CypB is not necessary for this physical binding (Figure 2c). CHOP is present in the cytosol under nonstressed conditions, and stress leads to the induction of CHOP and its accumulation in the nucleus.12 To confirm the interaction between CypB and CHOP in vivo, we examined the subcellular distribution of CypB and CHOP using confocal microscopy. AGS cells were transfected with HA-CypB and Flag-CHOP expression vector. The immunofluorescence analysis shows that CypB mainly colocalized with CHOP, but not completely, as shown in a merged image (Figure 2d, left panel). We repeated the colocalization experiment with endogenous CHOP and CypB in AGS cells under hypoxia. Consistently, the majority of endogenous CHOP colocalized with CypB under hypoxia (Figure 2d, right panel). To further study the interaction of CypB and CHOP, we used a set of deletion mutants of Myc-CHOP to coimmunoprecipitate with Flag-CypB in HEK293 QBI cells (Figure 2e). As shown in Figure 2f, wild-type CHOP (WT), CHOPΔBR (lacking whole-DNA-binding domain) and CHOPΔLZ (lacking leucine zipper domain) were tested to interact with CypB. However, CHOPΔN36 (lacking α-helix domain) failed to interact with CypB, suggesting that N-terminal region of CHOP containing α-helix domain is necessary for the interaction. These results were confirmed by performing the GST-pulldown assay (Figure 2g). Also, in order to address whether the N-terminal region of CHOP is necessary for its degradation by proteasome, degradation of CHOPΔN36 was monitored with increased expression levels of CypB. As shown in Figure 2h, the protein stability level of WT CHOP protein was decreased by CypB expression in a dose-dependent manner, but not CHOPΔN36, suggesting that N-terminal region of CHOP is necessary for its degradation. Next, we examined whether CypB and N-terminal region of CHOP are associated with polyubiquitination of CHOP. Consistent with the protein stability results, WT CHOP, CHOPΔBR and CHOPΔLZ were easily ubiquitinated; however, CHOPΔN36 was not readily ubiquitinated under the same conditions (Figure 2i). Taken together, our results suggest that CypB facilitates CHOP polyubiquitination through its binding to the N-terminal α-helix domain of CHOP.
CypB facilitates CHOP ubiquitination in a p300-dependent manner
Previously p300 was reported to be a crucial factor for the degradation of p53 as E4 ligase.13 The N-terminal region of CHOP necessary for its degradation was also essential for its interaction with p300.8 Therefore, we hypothesized that CypB might affect p300-dependent ubiquitination of CHOP. Given that p300 can act as an E4 for CHOP, the interaction between CHOP and p300 was analyzed by immunoprecipitation in hypoxia-induced HEK293 QBI cells treated with MG132 after HA-p300 and Flag-CypB overexpression. About two folds of CHOP was found to coimmunoprecipitate with HA-p300 after Flag-CypB overexpression compared with before, indicating that overexpression of CypB increased the interaction between CHOP and p300 (Figure 3a). We also repeated similar experiments in CypB knockdown. As shown in Figure 3b, CHOP binding to p300 was almost abolished in CypB knockdown in the presence of MG132. To further examine whether p300 affects the interaction between CypB and CHOP, we repeated the immunoprecipitation using p300 knockdown. Intriguingly, p300 knockdown significantly reduced CypB binding to CHOP in the presence of MG132 (Figure 3c). To further characterize the interactions among CypB, p300, and CHOP, coimmunoprecipitation experiment with endogenous CypB protein was performed in the presence of MG132 under hypoxia. Endogenous p300 was found to coimmunoprecipitate with endogenous CypB, suggesting that CypB and p300 physically interact with each other (Supplementary Figure 1A). Also, we repeated the coimmunoprecipitation experiment with endogenous p300. We successfully demonstrated that endogenous CHOP and CypB are precipitated with endogenous p300, suggesting that CypB, p300 and CHOP form a complex (Supplementary Figure 1B). Taken altogether, these results suggest that CypB and p300 cooperately bind to CHOP under hypoxia.

CypB enhances CHOP ubiquitination in a p300-dependent manner. (a and b) Coimmunoprecipitation of CHOP and p300 in HEK293 QBI cells treated with 20 μM of MG132 for 4 h. Overexpression of CypB increased the binding between CHOP and p300 (a), whereas knockdown of CHOP decreased their binding (b). (c) Coimmunoprecipitation of CypB and CHOP in HEK293 QBI cells treated with 20 μM of MG132 for 4 h. Knockdown of p300 diminished the binding between CypB and CHOP. (d and e) HEK293 QBI cells were cotransfected with Flag-CypB, HA-p300, Myc-CHOP, and HA-ubiquitin, and negative control siCon or siCypB, and protein extracts were immunoprecipitated. Ubiquitination of CHOP was measured with an anti-HA antibody. Cell lysates were immunoblotted with the indicated antibodies. (f and g) HEK293 QBI cells were cotransfected with Flag-CypB, Myc-CHOP, and HA-ubiquitin, and negative control siCon, sip300 or siCypB, and protein extracts were immunoprecipitated. Ubiquitination of CHOP was assessed as in (d). (h) A schematic representation of a model showing CypB regulates p300-dependent CHOP ubiquitination. Quantitative analysis of all the bands is shown on a bar graph using ImageJ program. Bars represent means±s.d.; *P<0.05
Consistently, the introduction of CypB markedly facilitated p300-mediated ubiquitination of CHOP (Figure 3d, compare lane 2 with lane 4), whereas the knockdown of CypB decreased p300-mediated CHOP ubiquitination (Figure 3e, compare lane 3 with lane 4). Together, these data suggest that CypB is responsible for facilitating the binding of CHOP and p300, leading to CHOP destabilization via p300-dependent CHOP ubiquitination. Consistently, p300 knockdown also impaired CypB-mediated CHOP ubiquitination (Figure 3f, compare lane 3 with lane 4). Depletion of both CypB and p300 almost prevented CHOP ubiquitination (Figure 3g, compare lane 2 with lane 4). These data together suggest that cooperative binding of CypB and p300 to CHOP are necessary for CHOP ubiquitination under hypoxia (Figure 3h).
Modulation of CHOP stability by CypB is not dependent on ATF4 under hypoxia
It has been reported that ATF4 is overexpressed under hypoxia,14 and that it can directly upregulate CHOP transcription through its binding to the CHOP promoter region.5, 6 Thus, we tested whether hypoxia increases ATF4 and CHOP expression in our cell lines. Both ATF4 and CHOP protein levels began to increase after 3 h in hypoxia (Figure 4a), and continuously increased for up to 12 h (experimental endpoint). However, Bcl-2 expression was suppressed in a time-dependent manner under hypoxia. Next, we transfected AGS cells with the WT CHOP promoter (CHOP-777) in order to assess whether hypoxia induces the CHOP promoter through ATF4-binding sites. As expected, the WT CHOP promoter (CHOP-777) showed a significantly increased luciferase activity under hypoxia, compared with normoxia (Figure 4b). To determine if CHOP is regulated by ATF4 at the transcriptional level in AGS cells, we transiently cotransfected AGS cells with both ATF4 expression plasmid and CHOP-777 plasmids and analyzed the luciferase activity of these transfected cells (Figure 4c). Transfection with both ATF4 and CHOP-777 plasmids showed higher luciferase activity compared with transfection with the CHOP-777 plasmids alone, indicating the important transcriptional role of ATF4 in CHOP expression. The successful knockdown of endogenous ATF4 protein expression by siRNA resulted in a decrease in luciferase activity of the CHOP promoter in hypoxia (Figure 4d, upper panel). Western blot showed that siRNA-ATF4 treatment resulted in a significant decrease in endogenous CHOP expression levels (Figure 4d, lower panel). These results demonstrate that hypoxia-induced CHOP transcription is mediated by ATF4 in AGS cells. Given that CypB preferentially binds to CHOP, we wondered whether CypB regulates ATF4-induced CHOP signaling. We investigated the effects of CypB knockdown on ATF4 induced-CHOP signaling in response to hypoxia. Depletion of CypB in AGS, HeLa, and HEK293 QBI cells significantly induced CHOP expression but not ATF4 under hypoxia, compared with the negative control (Figure 4e, compare lane 2 with lane 4). However, depletion of CypB did not lead to the upregulation of CHOP mRNA levels (Supplementary Figure 2C). To explore the role of CypB in ATF4-mediated CHOP upregulation in hypoxia, we overexpressed CypB in AGS cells and monitored the expression levels of ATF4 and CHOP under hypoxia. Overexpressed CypB significantly reduced the expression level of CHOP, but not that of ATF4, compared with pcDNA control vector (Figure 4f, compare lane 2 with lane 4). Further, overexpressed CypB in AGS cells did not reduce endogenous CHOP mRNA levels under hypoxia compared with pcDNA control vector (Supplementary Figure 2D). We also monitored the transcriptional effects of CypB on CHOP mRNA under hypoxia in ATF knockdown or CypB knockdown (Supplementary Figures 2E and F). Our data confirmed again that ATF4 but not CypB transcriptionally upregulates CHOP under hypoxia. In order to confirm that CypB modulates CHOP stability in an ATF4-independent manner under hypoxia, we performed the immunoprecipitation experiment (Figure 4g). As expected, overexpression of CypB reduced CHOP expression, whether ATF4 was overexpressed or not (Figure 4g, compare lanes 2 and 6 with lanes 4 and 8, respectively).

Modulation of CHOP stability by CypB is not dependent on ATF4 under hypoxia. (a) AGS cells were exposed to hypoxia for the indicated times. The expression levels of ATF4, CHOP and Bcl-2 protein were analyzed by western blot. (b and c) AGS cells were transiently transfected with the empty pGL3 vector, WT CHOP-777 promoter constructs (b) and ATF4 expression plasmids (c), and further incubated for 24 h under hypoxia conditions. Luciferase activity is expressed relative to that of normoxic cells. (d) AGS cells treated with negative control siCon or siATF4 were cultured under hypoxic conditions for 48 h and analyzed using the luciferase assay (upper panel). The expression of ATF4 and CHOP was analyzed by western blot with actin as a loading control (lower panel). (e) CypB knockdown enhanced hypoxia-mediated CHOP expression. AGS, HeLa, and HEK293 QBI cells transfected by negative control siCon or siCypB for 48 h. The levels of CHOP, ATF4, and CypB in cell lysates were monitered by immunoblotting. (f) AGS cells transfected with empty pcDNA and HA-CypB expression plasmids. After transfection, cells were exposed to hypoxia for 24 h and subjected to western blot (left panel). (g) Myc-CHOP and HA-CypB were expressed in pcDNA or ATF4-transfected HEK293 QBI cells by exposing to hypoxia conditions for 24 h. The level of CHOP in anti-Myc immunoprecipitate was analyzed by western blot. Quantitative analysis of all the bands is shown on a bar graph using ImageJ program. Bars represent means±s.d.; *P<0.05, **P<0.01 and #P<0.05
Hypoxia-mediated induction of CypB requires ATF6
Next, the CypB protein level was monitored in AGS cells under hypoxia. CypB protein expression was significantly increased under hypoxia (Figure 5a). A time-course experiment showed that CypB protein expression under hypoxia reached a maximum level after 12 h and was sustained until 24 h. These results were confirmed by immunocytochemical observation. Calnexin, a calcium-binding protein, was used as an ER marker. Increased CypB expression was shown in the ER lumen of AGS cells after a 24-h exposure to hypoxia (Figure 5b). Densitometric analysis showed that CypB expression was increased by more than twofold under hypoxia, suggesting a crucial role of CypB in hypoxia. The hypoxic condition also physiologically occurs in brain ischemia. We used bilateral common carotid artery occlusion (BCCAO) to monitor neuronal CypB in ischemia-induced hippocampus. The expression of both CypB and glucose-related protein (GRP) 78 in the dentate gyrus subfields was studied using immunohistochemical analysis (Figure 5c). GRP78 was used as an ER marker. One week after BCCAO, CypB protein expression had increased by 2.5-fold in the dentate gyrus subfields in ischemia-operated mice compared with sham-operated mice. GRP78 protein expression was also strongly increased in ischemia-operated mice. These results suggest that CypB expression is upregulated by hypoxia in many kinds of cells.

Hypoxia upregulates CypB expression. (a) AGS cells were exposed to hypoxia for the indicated times. The expression of CypB protein was analyzed by western blot. (b) After exposure to normoxic or hypoxic conditions for 24 h, cells were stained with anti-CypB (green) and the ER marker calnexin (red), followed by nuclear staining with DAPI (blue). Bars, 20 μm. The intensity of CypB staining in AGS cells was measured by the LSM510 confocal laser microscope software (right panel). (c) Immunohistochemical analysis of CypB and GRP78 in the hippocampus after brain ischemia. Immunostaining of CypB in the dentate gyrus subfields in the hippocampus in sham-operated or 7-min BCCAO mice was performed. The LSM510 confocal laser microscope software was used for quantitative analysis (right panel). Bars, 50 μm. The data are shown as the means±s.d. from three independent experiments. Quantitative analysis of western blot is shown using ImageJ program. Bars represent means±s.d.; *P<0.05
To determine whether CypB is regulated by p50ATF6 or spliced XBP1 (sXBP1) at the transcriptional level under hypoxia, we transiently cotransfected AGS cells with ATF6- or sXBP1-transcription factor expression plasmids and pGL3-CypB-250 plasmid and analyzed the luciferase activity of these transfected cells (Figure 6a, upper panel). p50ATF6 expression plasmids increased the luciferase activity in the transfected cells dose-dependently compared with cells transfected with the pcDNA control vector, but sXBP1 expression plasmids did not (Figure 6a, upper right panel). Western blot showed that CypB protein expression is increased by p50ATF6 in a dose-dependent manner, but not by sXBP1 expression plasmids (Figure 6a, lower panel).

ATF6 mediates hypoxia-induced transcriptional upregulation of CypB. (a) AGS cells were transfected with the pGL3-CypB-250 plasmids, active form-ATF6 (p50ATF6), and spliced XBP1 (sXBP1) expression plasmids. After transfection, the cells were assayed for luciferase activity (upper panel) and western blot (lower panel). (b) Effect of site-specific mutagenesis of ATF6 on the luciferase activity. AGS cells were transiently transfected with the WT and mutant form CypB-250 plasmids. After transfection, cells were exposed to normoxic or hypoxic conditions for 24 h and subjected to the luciferase assay. (c) ATF6 binds to the region of the CypB promoter containing the CRE. Protein–DNA complexes from AGS cells exposed to hypoxia for 48 h were subjected to ChIP analysis. Where indicated, non-specific IgG was used as a control in the IP and subjected to RT-PCR (left panel) or real-time PCR (right panel). (d) AGS cells treated with siCon or siATF6 were cultured under hypoxic conditions for 24 h and analyzed using the luciferase assay (left panel) and RT-PCR (right panel). (e) AGS cells were exposed to hypoxia for the indicated times in the absence or presence of 300 μM AEBSF for 4 h by real-time PCR. (f) AGS cells were transfected with the pGL3-CypB-250 plasmid, exposed to hypoxia for the indicated times in the absence or presence of 300 μM AEBSF for 4 h, and followed by the luciferase assay (left panel) and western blot (right panel). (g) AGS cells were exposed to hypoxia for 48 h in the absence or presence of 300 μM AEBSF for 4 h, and cell death rate was evaluated using the trypan blue exclusion assay and expressed as a percentage of the stained cells. (h) AGS cells were exposed to hypoxia for 48 h and incubated in the absence or presence of 300 μM AEBSF for 4 h, and binding of ATF6 to the element A (irrelevant site) and B (CRE site) regions of the CypB promoter was analyzed by ChIP. Quantitative analysis of all the bands is shown using ImageJ program. Bars represent means±s.d.; *P<0.05, **P<0.01
To determine whether the ATF6 binding site is associated with hypoxia-induced transcriptional upregulation, the ATF6 binding site was mutated from ‘cgtgg’ to ‘cacag’ by site-directed mutagenesis in the pGL3-CypB-250 plasmid (Figure 6b). The mutated ATF6 binding site markedly abolished hypoxia-induced transcriptional activation of CypB promoter, indicating that ATF6 has a crucial role in hypoxia-induced transcriptional activation of CypB. To confirm these results, the binding of ATF6 to the CypB promoter under hypoxia was demonstrated by chromatin immunoprecipitation (ChIP) assays using an ATF6 antibody. ChIP assays with immunoglobulin G (IgG) antibody as a nonspecific control did not result in precipitation. After isolation of cross-linked chromatin from cells incubated with or without hypoxia, endogenous ChIP assays in AGS cells using RT-PCR and real-time PCR analysis demonstrated that ATF6 was recruited to the region containing the ATF6 binding site (element B) but not to an irrelevant site (element A; Figure 6c) in the CypB promoter region under hypoxia. These results were further confirmed by ATF6-knockdown experiments using siRNA under hypoxia. RT-PCR experiments demonstrated the successful siRNA-mediated knockdown of ATF6 (Figure 6d, right panel). Successful knockdown of endogenous ATF6 by siRNA abolished hypoxia-induced transcriptional activation of CypB (Figure 6d, left panel). These results indicate that hypoxia-mediated CypB transcription activation requires ATF6.
To confirm the crucial roles of the ATF6 pathway in hypoxia, a selective inhibitor of ATF6, 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), was used. AEBSF is a serine protease inhibitor and prevents ER stress-induced cleavage of p90ATF6.15 The real-time PCR analysis also showed that both ATF6 mRNA (Figure 6e, left panel) and CypB mRNA levels were decreased by AEBSF under hypoxia (Figure 6f, right panel). Consistently, hypoxia-induced CypB luciferase activity was decreased in the presence of AEBSF, compared with the absence of AEBSF (Figure 6f, left panel). Western blot showed that AEBSF efficiently blocks p90ATF6 cleavage and inhibits hypoxia-mediated induction of CypB (Figure 6f, right panel). Interestingly, downregulation of CypB by AEBSF increased hypoxia-induced cell death compared with the absence of AEBSF (Figure 6g). Also, ChIP assay demonstrated that AEBSF abrogated the hypoxia-stimulated ATF6 recruitment to the CypB promoter (element B), but not to an irrelevant site (element A; Figure 6h). Taken together, these results suggest a crucial role for ATF6 in the induction of CypB under hypoxia.
Overexpressed CypB inhibits hypoxia-induced apoptosis via ER stress-dependent pathway
To explore the role of CypB in hypoxia-induced cell death, we monitored DNA fragmentation by the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay. As expected, CypB expression inhibited hypoxia-induced DNA fragmentation compared with pcDNA control vector (Figure 7a, upper panel). As shown by western blot analysis, the expression of cleaved caspase-3 and anti-poly (ADP ribose) polymerase (PARP) was almost suppressed by overexpressed CypB under hypoxia (Figure 7a, lower panel). These observations were further confirmed by flow cytometry (FACS) analysis (Figure 7b), the clonogenic assay (Figure 7c) and CypB knockdown experiments (Figure 7d). In order to examine whether CypB knockdown may cause ER stress-induced apoptosis under hypoxia, several apoptosis markers were monitored. Knockdown of CypB increased the expression levels of cleaved caspase3, PARP, Bax, GRP78, p-eIF2α, and decreased Bcl-2 expression compared with negative control siRNA in hypoxia (Figure 7e). Also, we monitored CHOP activation in CypB knockdown-induced apoptosis under hypoxia. CHOP was easily detected in the nucleus of apoptotic cells in CypB knockdown by RNA interference, but not in the cells with the negative control siRNA in hypoxia (Figure 7f). Taken together, these results indicate that CypB knockdown increased hypoxia-induced apoptosis through an ER stress-dependent pathway.

Overexpression of CypB suppresses hypoxia-induced apoptosis by ER stress-dependent pathways. (a) AGS cells transfected with HA-CypB (4 μg), were exposed to hypoxia for 48 h. Cells were fixed, permeabilized, and followed by the TUNEL assay. TUNEL-positive cells were quantified and plotted (upper panel). The expression levels of HA-CypB, cleaved-caspase3 and cleaved-PARP protein were analyzed by western blot (lower panel). (b) AGS cells were transfected with empty pcDNA (4 μg) and HA-CypB (4 μg), and then exposed to hypoxia for 48 h. Cells were harvested and subjected to propidium iodide staining. The apoptosis rate was measured by sub-G1 DNA contents using FACS. (c) Attenuation of hypoxia-induced apoptosis by overexpression of CypB (4 μg) was determined by the clonogenic assay. Surviving fractions were determined based on plating efficiencies. (d) Knockdown efficiency of siCypB was determined by western blot. Cell death rate was evaluated using the trypan blue exclusion assay and expressed as a percentage of the stained cells. (e) AGS cells treated with negative control siCon or siCypB were cultured under hypoxia for 48 h and subjected to western blot analysis for the evaluation of proteins involved in apoptotic cell death. The expression levels of CypB, cleaved-caspase 3, cleaved-PARP, Bax, Bcl-2, GRP78, and p-elF2α were analyzed. Actin was used as a loading control. (f) CHOP activation in CypB knockdown-induced apoptosis under hypoxia. After exposure to hypoxic conditions for 48 h, cells were stained with anti-CypB (green), anti-CHOP (red) and followed by nuclear staining with DAPI (blue). Bars, 20 μm. (g) AGS cells transfected with negative control siRNA and CypB siRNA for 48 h were exposed to normoxic or hypoxic conditions for 48 h and the double-knockdown efficiency of siCypB and siCHOP was determined by the trypan blue exclusion assay and expressed as a percentage of the stained cells. (h) Schematic model showing that ATF6-mediated upregulated CypB facilitates p300-dependent CHOP ubiquitination under hypoxia. Bars represent means±s.d.; *P<0.05, **P<0.01 and #P<0.05
Finally, in order to investigate whether CypB and CHOP are controlling cell death on a linear pathway, we performed a double knockdown of CypB and CHOP in AGS cells (Figure 7g). Each knockdown efficiency was checked by immunoblots (Supplementary Figure 3). The double knockdown cells were incubated under hypoxia for 48 h and subjected to the trypan blue exclusion assay. CypB knockdown showed a significant increase in apoptotic cells compared with control siRNA-transfected cells under hypoxia. However, CHOP knockdown showed a significant decrease in apoptotic cells, compared with control siRNA-transfected cells under hypoxia. These results suggest that CypB has an anti-apoptotic role, whereas CHOP seems to function as a pro-apoptotic factor in our cell system. The double knockdown showed a similar level of cell death compared with CHOP knockdown under hypoxia, but a significant decrease compared with CypB knockdown under hypoxia. Our data suggest that CypB and CHOP control hypoxia-induced cell death on a linear pathway.
Discussion
The transcription factor CHOP is one of the best-characterized pro-apoptotic molecules accounting for ER stress-induced cell death. However, little is known about the interaction of CypB and CHOP under hypoxia. Here, we demonstrated that CypB and p300 cooperatively interacts with CHOP, leading to ubiquitination of CHOP and inhibition of hypoxia-induced apoptosis.
In the current study, we found that hypoxia significantly upregulated CypB mRNA expression in a time-dependent and an ATF6-dependent manner (Figure 6). This is inconsistent with the results of a previous report showing that HIF-1α transcriptionally upregulates CypB under hypoxia.16 Thus, it is possible that hypoxia-mediated CypB gene induction is due, at least in part, to HIF-1α activation. However, under the conditions employed in this study, a site-directed mutagenesis in the ATF6 binding site prevented hypoxia-induced upregulation of CypB. CypB upregulation in ATF6 knockdown cells by siATF6 was significantly impaired. Finally, we could demonstrate that AEBSF, an ATF6 inhibitor, could not induce CypB expression under hypoxia (Figure 6). Also, these results are consistent with those of previous studies showing that HIF-1α activation is minimal under very low oxygen concentrations,17 such as 0.1% used in the current study. Brain ischemia has also been reported to activate the eukaryotic initiation factor 2α kinase PERK, an ER-stress sensor.18 Interestingly, CypB was also induced by brain ischemia (Figure 5). Furthermore, overexpression of CypB significantly prevented hypoxia-induced cell death, whereas the cell death was augmented in the CypB-knockdown cells (Figure 7).
Here, we showed that ubiquitination of CHOP was regulated by cooperative binding of CypB and p300 to the N-terminal portion of CHOP. CHOPΔN36 was not readily ubiquitinated, indicating that the region up to a.a 35 of CHOP was essential for its degradation. The potential mechanism underlying the cytoprotective role of CypB in hypoxia is summarized in Figure 7h. Hypoxia induces CypB and CHOP through ATF6 and ATF4, respectively. Association of CypB, CHOP, and p300 promotes the ubiquitination of CHOP, a pro-apoptotic molecule. This process suppresses hypoxia-induced cell death.
Chaperone function has been traditionally linked to the folding and assembly of proteins.19, 20, 21 However, in recent years multiple chaperone-assisted degradation pathways have also emerged, including chaperone-assisted proteasomal degradation, chaperone-assisted selective autophagy, and chaperone-mediated autophagy.22 We also have showed in detail a novel CypB–CHOP complex that enhances the rate of ubiquitination of CHOP. Although the associations between molecular chaperones and the ubiquitin-proteasome system seem to be critical for cytoprotection in response to ER stress, the relationship between chaperone-proteasome interaction and cellular physiological events remains to be explored in detail. Furthermore, many more studies are required to fully understand the exact role of CypB in regulating the stability of other possible binding proteins. Nonetheless, taken together, these results make CypB an attractive therapeutic target, the inhibition of which could potentially upregulate the expression of CHOP and other pro-apoptotic proteins to retard the growth of human tumor cells.
Materials and Methods
Reagents and antibodies
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS) and Roswell Park Memorial Institute (RPMI) 1640 medium were purchased from Lonza (Walkersville, MD, USA). Crystal violet solution, actinomycin D, CHX, Flag-tag and propidium iodide were purchased from Sigma-Aldrich (St Louis, MO, USA). Lipofectamine 2000 reagents were purchased from Invitrogen (Carlsbad, CA, USA). Antibodies for caspase3, CHOP and phospho-eIF2α (Ser51) were purchased from Cell Signaling Technology (Danvers, MA, USA). 4′,6′-Diamidino-2′-phenylindole dilactate, AEBSF, anti-glucose-related protein 78/Bip, anti-actin, anti-Bax, anti-Bcl-2, PARP, anti-α-actinin, anti-p300, IgG, HA-tag, protein G/A bead, anti-GST and ECL Western blotting kit was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cyclophilin B, calnexin, ATF6, and ATF4 were purchased from Abcam (Cambridge, UK). Anti-Myc monoclonal antibody (9E10) was from Roche Applied Science (Mannheim, Germany). Ubiquitin and MG132 were purchased from Assay Designs (Ann Arbor, MI, USA). Apo-BrdU In Situ DNA Fragmentation Assay Kit was purchased from BioVision (Mountain View, CA, USA). Other chemical reagents were obtained from Sigma-Aldrich.
Plasmid constructions
A cDNA encoding the C-terminal 216 amino acids of WT CypB and PPIase-defective CypB R95A mutant was subcloned into the pcDNA vector (Invitrogen) between HindIII and EcoRV sites for protein expression, which tagged the protein with an HA-tag at the C-terminal end. Flag-CHOP, Flag-ATF4, Flag-XBP1 and Flag-CypB were subcloned into pCMV-Taq 2B flag vector using BamHI and XhoI restriction sites. GST-CypB and CypB series region reporter plasmids have been described previously.10 The active-ATF6 expression vector was kindly provided by Dr. Kyung Ho Lee (Konkuk University, Seoul, Korea). The human CHOP promoter reporter constructs were kindly provided by Dr. P Fafournoux (Unité Nutrition Humaine, INRA de Theix, Champanelle, France). HA-p300 and HA-ubiquitin expression vectors were kindly provided by Dr. J Ha (Kyung Hee University, Seoul, Korea). The plasmids pcDNA3.1-Myc-CHOP, ΔBR, ΔLZ, and ΔN36 were kindly provided by Dr. Hidetoshi Hayashi (Nagoya City University, Nagoya, Japan). All constructs were verified by sequencing.
Cell culture and in vitro hypoxia model
AGS (human gastric carcinoma) cells were maintained in RPMI 1640 supplemented with 10% heat-inactivated FBS. HeLa (human cervix adenocarcinoma) and HEK293 QBI cellswere cultured in DMEM supplemented with 10% FBS in humidified air containing 5% CO2 at 37 °C. HEK293 QBI cell line is a superior subclone of HEK 293 cells, harboring the E1A and E1B regions of the adenoviral genome, and complementing in trans the deletion of the E1 region in the recombinant Adenovirus(Qbiogene, Livingstone, UK). The cells were incubated under either normoxia (20% O2) or hypoxic conditions (0.1% O2). Cultured AGS cells were subjected to hypoxic conditions at 37 °C for the time periods indicated, whereas controls were left in normoxic conditions at 37 °C for the same time periods.
Mouse ischemia model and immunohistochemistry
The experiments using mice were treated in accordance with the principles and procedures of the Kyung Hee University Guidelines for the Care and Use of Laboratory Animals. All protocols were approved by the Institutional Animal Care and Use Committee of Kyung Hee University. Animals were subjected to global ischemia by temporary BCCAO (7 min) or to sham surgery, followed by reperfusion, as described previously.23 The sections were analyzed using an LSM510 confocal laser microscope (Carl Zeiss, Oberkochen, Germany).
RT-PCR and real-time PCR
Total cellular RNA was extracted from cells using the TRIzol reagent (Invitrogen). A cDNA was synthesized from 2 μg of total RNA using M-MLV reverse transcriptase (Fermentas, Hanover, MD, USA). Specific primers for RT-PCR: CypB (sense) 5′-AATTCCATCGTGTAATCAAGGACTT-3′ and (anti-sense) 5′-TCTTGACTGTCGTGATGAAGAACT-3′; ATF6 (sense) 5′-TTTAAGCTTATGGGGGAGCCGGCTGGG-3′ and (antisense) 5′-CCGCTCGAGCTATTGTAATGACTCAGGG-3′; CHOP (sense) 5′-GCGCATGAAGGAGAAAGAAC-3′ and (antisense) 5′-CCAATTGTTCATGCTTGGTG-3′; β-actin (sense) 5′-GTACTTGCGCTCAGGAGGAG-3′ and (antisense) 5′-TCGTGCGTGACATTAAGGGG-3′. PCR products were analyzed by agarose gel electrophoresis and visualized using ethidium bromide. The signals were quantitated using Image J software (National Institutes of Health (NIH), Bethesda, MD, USA). Real-time PCR was performed using an ABI prism 7300 Sequence Detection System (Applied Biosystems, Branchburg, NJ, USA) with SYBRGreen PCR Master Mix (Applied Biosystems). The PCR reaction was carried out for 40 thermal cycles. Expression of the target gene was analyzed by an absolute quantification method and normalized using β-actin levels.
Luciferase assay
The CypB promoter sequence was analyzed using Genomatrix MatInspector (http://www.genomatix.de). One ERSE candidate was located at −222 bp, approximately −209 bp from the CypB ORF. AGS cells were transfected with 0.8 μg of the pGL3 basic-derived plasmids together with the internal control plasmid, pCMV-β-galactosidase (Promega, Madison, WI, USA). Luciferase and β-gal activities were measured using 50 μl of each cell lysate using a fluorescence microplate reader, and the luciferase activity was normalized on the basis of β-galactosidase values. Transfection experiments were performed in triplicates and repeated at least three times.
GST pull-down assay
GST fusion proteins, GST, and GST-CypB were expressed in an Escherichia coli DH5α strain and affinity purified on glutathione-sepharose 4B beads. Isopropyl-1-thio-β-D-galactopyranoside was added in a growing exponential bacterial culture to a final concentration of 1 mM and incubated for 4 h at 37 °C. Whole-cell lysates from HEK293 QBI cells were incubated with total cells extracted and treated for 24 h. After extensive washes, SDS-PAGE sample buffer was added to the beads, boiled, and subjected to SDS-PAGE. Fractionated proteins were visualized by western blot.
Western blot and coimmunoprecipitation
The cells were washed twice with cold phosphate-buffered saline (PBS) on ice and harvested by scraping with a rubber policeman. The cells were pelleted by centrifugation at 4 °C and resuspended directly into a lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% Igepal CA-630, 2 mM EDTA, 10 mM NaF, 2 mM Na3VO4, and 0.01% protease inhibitor cocktail). Cell lysates were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. After blocking in 5% skimmed milk and tris-buffered saline with 0.1% Tween-20, signals were detected and analyzed by a Kodak X-OMAT 2000 image analyzer (Rochester, NY, USA). For immunoprecipitation experiments, cell lysates were precleared with protein G/A beads and subsequently incubated for 1–2 h with protein G/A beads covalently coupled with anti-CypB, anti-CHOP, and anti-p300. Immune complexes were washed four times with cell extraction buffer. Eluted samples or whole-cellular lysates were resolved by SDS-PAGE and proteins were detected by western blot using the indicated antibodies. Densitometric analysis was performed using ImageJ software. Unless otherwise specified, actin protein was immunoblotted in order to normalize the quantity of sample protein.
Ubiquitination assay
The HEK293 QBI cells were transiently cotransfected with indicated plasmids and were treated with 20 μM MG132 for 4 h before harvesting. Cells were lysed with lysis buffer and immunoprecipitated with anti-CHOP or HA antibodies. The ubiquitination reaction mixtures were collected by centrifugation and washed three times with lysis buffer. The samples were then subjected to SDS-PAGE followed by western blot.
Site-directed mutagenesis
Site-directed mutagenesis of the CypB promoter (pGL-250) region was performed using a QuickChange Site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) according to the instruction manual. The pGL3-250/CypB vector was used as a template DNA and PCR based site-directed mutagenesis were as follows: ATF6 binding site (sense 5′-GAGCAATAACACAGCAACCACCC-3′, and anti-sense 5′-CTCGTTATTGTGTCGTTGGTGGG-3′). The underlined letters represent the mutated sequence for the site-directed mutagenesis. PCR was performed using two antiparallel primers with the required nucleotide substitutions and pGL3-250/CypB plasmid as a template. The PCR product was then treated with DpnI endonuclease. The sequences of the mutation constructs were confirmed by DNA sequencing.
ChIP assay
ChIP assay was conducted using the ChIP assay protocol (Upstate, Lake Placid, NY, USA) according to the manufacturer’s instructions. Primers were as follows: For element A, forward, 5′-GTAACTATTCCTGACCTCCGGAA-3′, reverse, 5′-CAGTACTGAGTGCCCAACAAGTC-3′; for element B, forward, 5′-CATGCCGGGGCTTGTGGTCTTCTCGG-3′, reverse, 5′-GCGCCGTTTCCCCCCTCCGGCTC-3′).
RNA interference
Double-stranded RNA duplexes corresponding to human CypB (5′-CAAAAACAGUGGAUAAUUU-3′), human p300 (5′-GCACAAAUGUCUAGUUCUUTT-3′), human ATF4 (5′-GCCUAGGUCUCUUAGAUGA-3′), human ATF6 (5′-CACAACAGAGUCUCAGG-3′), human CHOP (5′-CAAUUGUUCAUGCUUGGUGUU-3′), and control siRNA (5′-GCGCGCUUUGUAGGAUUCG-3′) were designed. They were purchased from Dharmacon (Chicago, IL, USA). Each well was incubated for 48 h with 100 pmol using lipofectamine 2000 reagents (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations. The cells were then washed off the plates and the transfected cells were placed in a serum-free medium, after which they were subjected to various treatments.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed as described previously.16 Images were taken using the laser-scanning confocal microscope. For quantification, cells for each condition were analyzed in three sets.
Clonogenic assay
Long-term survival was determined by a colony-forming assay. A total of 1 × 103 cells were plated onto 60-mm dishes. At 24 h after plating, cells were transfected with either an empty vector (pcDNA3) or HA-CypB expression vector and incubated for 2 days under hypoxia (0.1% O2) conditions. Cell monolayers were rinsed twice and then fresh medium was added. After 10 days, the colonies were fixed with methanol and stained with 0.1% crystal violet solution. After 30 min, the absorbance of the reaction mixtures was measured at 595 nm using a fluorescence microplate reader (Bio-Rad, Hercules, CA, USA).
TUNEL assay
The TUNEL assay was conducted using the Apo-BrdU In Situ DNA Fragmentation Assay Kit following the protocol of the supplier. The TUNEL reaction was performed according to the manufacturer’s instructions. The number of apoptotic cells was expressed as a percentage of the total cells counted from five random fields for each cover slip.
Stable cell line development
For the development of pcDNA3 and WT CypB, AGS and HeLa cells were transfected with pcDNA3 and HA-tagged CypB. Two days post transfection, cells were selected with 1 mg/ml G418 sulfate until distinct colonies emerged. Colonies were propagated and examined for CypB and HA expression levels.
Apoptosis assay
For FACS analyses of cell cycle and apoptosis, cells were fixed in 70% (v/v) ethanol in PBS and then stored at 4 °C. The fixed cells were washed in PBS, incubated with 1 ml of a solution containing RNase A (50 mg/ml) and propidium iodide (50 mg/ml) solution for 30 min at room temperature. At least 1 × 104 cells were collected and analyzed by flow cytometry (Beckman-Coulter, Brea, CA, USA). Percentages of cells in sub-G1 were calculated with Multicycle for Windows software (Beckman-Coulter). Apoptotic cell death was determined by the trypan blue exclusion assay. The hypoxic cells were trypsinized and then centrifuged at 1000 × g for 5 min. Both dead cells floating in the medium and the cells attached to the plate were recovered and resuspended in 500 μl medium. The trypan blue exclusion assay was performed using the Vi-cellTM XR (Beckman-Coulter).
Statistical analysis
The results were expressed as means±s.d. from at least three independent experiments. Statistical analyses were conducted using Student’s t-tests. By convention, a P-value of <0.05 was considered statistically significant.
Abbreviations
- CypB:
-
cyclophilin B
- PPIase:
-
peptidyl-prolyl cis–trans isomerase
- ER:
-
endoplasmic reticulum
- GRP:
-
glucose-regulated protein
- UPR:
-
unfolded protein response
- IRE1α:
-
inositol-requiring transmembrane kinase and endonuclease 1α
- PERK:
-
RNA-dependent protein kinase-like ER kinase
- ATF6:
-
activating transcription factor 6
- ATF4:
-
activating transcription factor 4
- eIF2α:
-
eukaryotic translation initiation factor 2α
- CHOP:
-
CCAAT/enhancer-binding protein-homologous protein
- JNK:
-
c-Jun NH2-terminal kinase
- TUNEL:
-
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- CHX:
-
cycloheximide
- BCCAO:
-
bilateral common carotid arteries occlusion
References
Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 1998; 12: 982–995.
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ . Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001; 21: 1249–1259.
Luethy JD, Holbrook NJ . Activation of the gadd153 promoter by genotoxic agents: a rapid and specific response to DNA damage. Cancer Res 1992; 52: 5–10.
Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol 1996; 16: 4273–4280.
Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ . Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J 1999; 339: 135–141.
Averous J, Bruhat A, Jousse C, Carraro V, Thiel G, Fafournoux P . Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J Biol Chem 2004; 279: 5288–5297.
Friedman AD . GADD153/CHOP, a DNA damage-inducible protein, reduced CAAT/enhancer binding protein activities and increased apoptosis in 32D c13 myeloid cells. Cancer Res 1996; 56: 3250–3256.
Ohoka N, Hattori T, Kitagawa M, Onozaki K, Hayashi H . Critical and functional regulation of CHOP (C/EBP homologous protein) through the N-terminal portion. J Biol Chem 2007; 282: 35687–35694.
Gupta S, Read DE, Deepti A, Cawley K, Gupta A, Oommen D et al. Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell Death Dis 2012; 3: e333.
Kim J, Choi TG, Ding Y, Kim Y, Ha KS, Lee KH et al. Overexpressed cyclophilin B suppresses apoptosis associated with ROS and Ca2+ homeostasis after ER stress. J Cell Sci 2008; 121: 3636–3648.
Hattori T, Ohoka N, Inoue Y, Hayashi H, Onozaki K . C/EBP family transcription factors are degraded by the proteasome but stabilized by forming dimer. Oncogene 2003; 22: 1273–1280.
Ron D, Habener JF . CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev 1992; 6: 439–453.
Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 2003; 300: 342–344.
Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 2004; 24: 7469–7482.
Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y et al. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J Biol Chem 2003; 278: 31024–31032.
Kim Y, Jang M, Lim S, Won H, Yoon KS, Park JH et al. Role of cyclophilin B in tumorigenesis and cisplatin resistance in hepatocellular carcinoma in humans. Hepatology 2011; 54: 1661–1678.
Jiang BH, Semenza GL, Bauer C, Marti HH . Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol 1996; 271: C1172–C1180.
Kumar R, Azam S, Sullivan JM, Owen C, Cavener DR, Zhang P et al. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J Neurochem 2001; 77: 1418–1421.
Bukau B, Horwich AL . The Hsp70 and Hsp60 chaperone machines. Cell 1998; 92: 351–366.
Frydman J . Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem 2001; 70: 603–647.
Young JC, Agashe VR, Siegers K, Hartl FU . Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol 2004; 5: 781–791.
Kettern N, Dreiseidler M, Tawo R, Hohfeld J . Chaperone-assisted degradation: multiple paths to destruction. Biol Chem 2010; 391: 481–489.
Wu C, Zhan RZ, Qi S, Fujihara H, Taga K, Shimoji K . A forebrain ischemic preconditioning model established in C57Black/Crj6 mice. J Neurosci Methods 2001; 107: 101–106.
Acknowledgements
This study was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health, Welfare, and Family Affairs, Republic of Korea (A092125) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (20120009380).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Jeong, K., Kim, H., Kim, K. et al. Cyclophilin B is involved in p300-mediated degradation of CHOP in tumor cell adaptation to hypoxia. Cell Death Differ 21, 438–450 (2014). https://doi.org/10.1038/cdd.2013.164
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.164
Keywords
This article is cited by
-
High-fat diet induced cyclophilin B enhances STAT3/lncRNA-PVT1 feedforward loop and promotes growth and metastasis in colorectal cancer
Cell Death & Disease (2022)
-
Parkin regulation of CHOP modulates susceptibility to cardiac endoplasmic reticulum stress
Scientific Reports (2017)
-
Cycloheximide promotes paraptosis induced by inhibition of cyclophilins in glioblastoma multiforme
Cell Death & Disease (2017)

{kind=link}
{kind=link}
{kind=link}